
Reklama
Spis treści
Mimo dobrych wyników efektywności w zakresie utraty wagi przez rozwijaną w II fazie cząsteczkę VK2735 akcje Viking Therapeutics straciły jednego dnia 42%. Inwestorom nie spodobało się to, że 38% pacjentów zaprzestało terapii z powodu działań niepożądanych. FDA zacznie codziennie publikować dane ze swojego systemu zgłaszania zdarzeń niepożądanych. Rezdiffra, lek Madrigal Pharmaceuticals, służący terapii MASH, w końcu znalazł się na salonach unijnych po decyzji Komisji Europejskiej - najpierw trafi do niemieckich pacjentów.
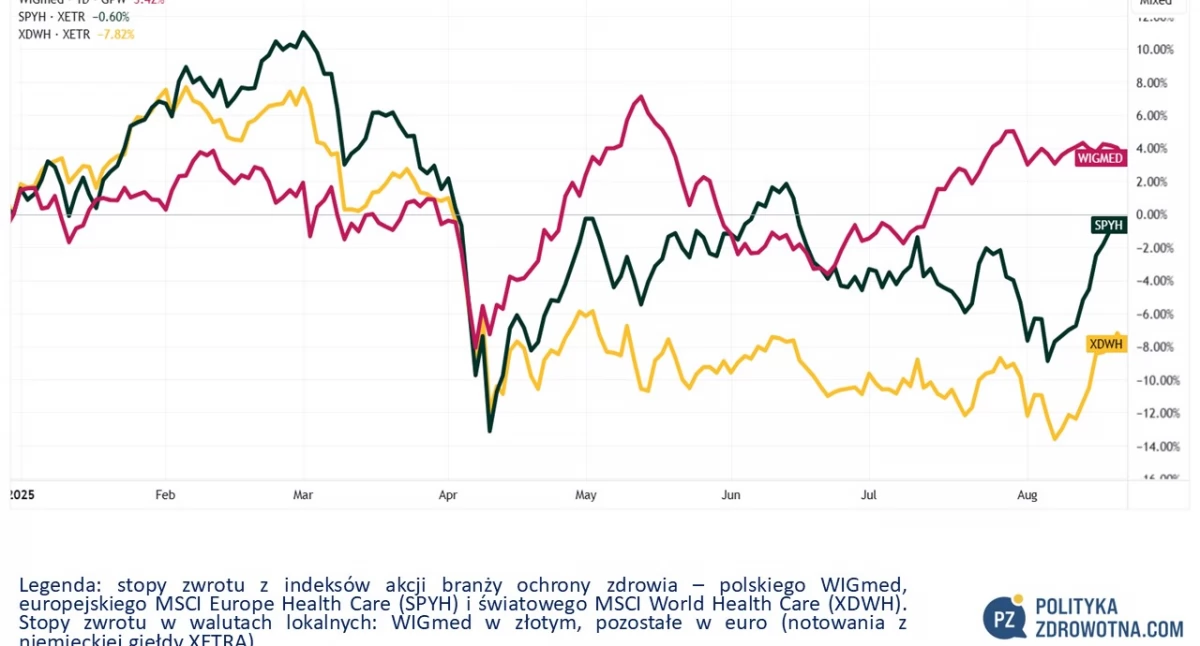
Polski WIGmed minimalnie zmienił się w ostatnim tygodniu giełdowym (-0,3%). Na plusach, i to sporych, zakończyły piątek zagraniczne indeksy akcji. Europejski MSCI Europe Health Care (SPYH) zyskał 3,4%, co okazało się drugim tygodniem z rzędu, gdy poszedł w górę o co najmniej 3%. Dobrze też radził sobie MSCI World Health Care (XDWH), który zwyżkował 2,0%.
Licząc od początku tego roku rodzimy WIGmed jako jedyny z analizowanych przez nas indeksów jest nad kreską, ale za sprawą ponadprzeciętnego zachowania się akcji europejskich w minionych dwóch tygodniach jego przewaga topnieje.
Reklama
Źródło: TradingView
W gronie akcji światowych z czołowej dwudziestki indeksu MSCI Global Health Care najbardziej wyróżniły się na plus walory Novo Nordisk (+8,7%), Mercka (+3,8%) i Roche (+3,4%). Indeks wciąż wykazywał swoją siłę, ponieważ tylko trzech emitentów zamknęło tydzień w rejonie ujemny. Te jednostkowe przypadki to: Gilead Sciences (-3,0%), Intuitive Surgical i Amgen (obie po -0,9%).
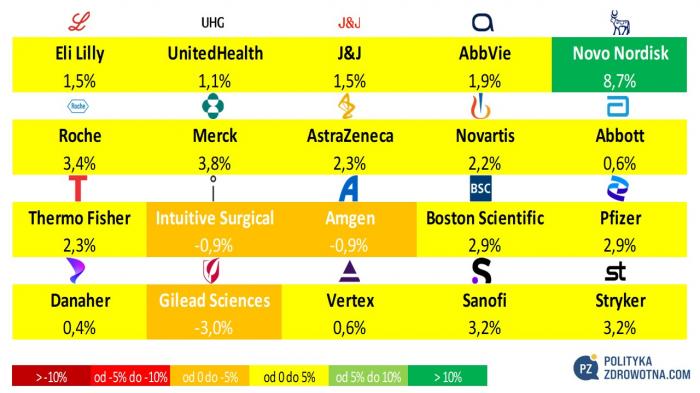
Źródło: opracowanie własne na podstawie stooq.pl
Polska nadal odstawała od reszty świata pod względem koniunktury, choć i u nas dało się znaleźć spółki, które odnotowały dodatnią stopę zwrotu. NanoGroup zyskało 5,0%, Scope Fluidics 4,1%, zaś Synektik 2,3%. Największymi przegranymi ostatniego tygodnia stały się PolTREG (-7,4%), Genomtec (-6,1%) oraz ex-aequo Voxel, Ryvu i Enel-Med (-2,7%).
Reklama
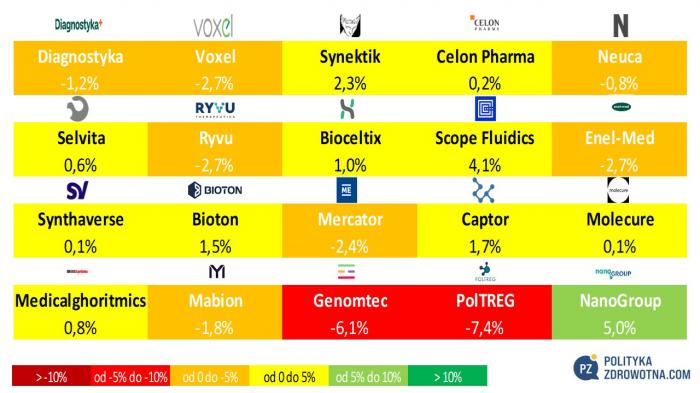
Źródło: opracowanie własne na podstawie stooq.pl
FDA idzie w kierunku większej przejrzystości
W końcu stanie się to, czego od dawna oczekiwało w szczególności środowisko pacjentów, praktyków medycyny i farmacji oraz inwestorów. FDA pod naciskiem amerykańskiego Departamentu Zdrowia i Opieki Społecznej (HHS) ukierunkowanym na radykalną przejrzystość podała w piątek, że zacznie codziennie publikować i aktualizować dane ze swojego systemu zgłaszania zdarzeń niepożądanych (FAERS).
FAERS to podstawowa baza danych FDA służąca do gromadzenia i analizowania raportów o zdarzeniach niepożądanych, poważnych błędach w stosowaniu leków i skargach dotyczących jakości produktów dla leków na receptę i terapeutycznych leków biologicznych, zawierająca raporty przedkładane przez pracowników służby zdrowia, konsumentów i producentów. Dostęp do niej można uzyskać pod tym linkiem: https://www.fda.gov/drugs/fdas-adverse-event-reporting-system-faers/fda-adverse-event-reporting-system-faers-public-dashboard.
Reklama
„Zgłaszanie zdarzeń niepożądanych powinno być szybkie, bezproblemowe i przejrzyste" - powiedział Marty Makary, komisarz urzędu.
"Ludzie, którzy poruszają się po niezgrabnych rządowych stronach internetowych zgłaszających zdarzenia niepożądane, nie powinni czekać miesiącami, aż te informacje zostaną upublicznione. Zamykamy ten okres oczekiwania i będziemy nadal usprawniać proces od początku do końca" - dodał.
Dlaczego wcześniej taka procedura nie została wdrożona w życie urzędnik nie wyjaśnił, choć postulaty w tej sprawie wielokrotnie zgłaszane były publicznie.
Reklama
Akcje Vikinga straciły we wtorek 42%
Inwestorzy z coraz tłoczniejszego segmentu terapii nadwagi/otyłości nie wybaczają nawet najdrobniejszych pomyłek, o czym świadczy gigantyczna przecena akcji Viking Therapeutics, które solidnie spadły po tym, gdy spółka poinformowała, że wielu pacjentów przyjmujących doustną postać peptydu koagonistycznego GLP-1/GIP VK2735 przerwało leczenie z powodu zdarzenia niepożądanego.
Początkowo w procesie odkrywczym wszystko zapowiadało się nad wyraz obiecująco. W badaniu I fazy klinicznej VK2735 odnotowano 14% wskaźnik nudności i, w co nie chciało aż się wierzyć, zerowy wskaźnik przypadków wymiotów. W II etapie badań było już dużo gorzej - wskaźniki obu zmiennych wyniosły odpowiednio 60% i 35%.
Reklama
Wydaje się, że problemy z tolerancją przysłoniły inwestorom dobrą efektywność kandydata na lek. Uczestnicy badania II fazy, którzy otrzymali 120-mg dawkę VK2735, zauważyli 12,2% zmniejszenie średniej masy ciała w stosunku do wartości wyjściowej po 13 tygodniach w porównaniu z 1,3% w przypadku placebo. To był dobry rezultat, gdyż analitycy Wall Street deklarowali, że uznaliby wskaźnik na poziomie 10% jako ponadprzeciętny.
Czarę goryczy przelał jednak fakt, że po podaniu najwyższej dawki leku Vikinga 38% pacjentów przedwcześnie przerwało leczenie, w porównaniu z 18% w przypadku placebo. Firma wskazała, że 98% zdarzeń niepożądanych związanych z leczeniem w badaniu VK2735 zostało zaklasyfikowanych jako łagodne lub umiarkowane pod względem nasilenia, a 99% miało charakter żołądkowo-jelitowy.
Reklama
Jared Holz, analityk Mizuho, zwrócił uwagę, że pacjenci w badaniu Vikinga odstawili VK2735 w tak szybkim tempie - w ciągu 13 tygodni, co może nie wróżyć dobrze przyszłości rozwojowej cząsteczki.
Jak Vanda dzielnie walczyła z niemocą
Pozostajemy w kręgu litery „V”. Viking nie ma wiele wspólnego z Vandą, bo legendarna krakowska Wanda ponoć nie chciała Niemca, dlatego odebrała sobie życie, topiąc się w Wiśle. Prawdziwa Vanda jest dużo bardziej pragmatyczna niż romantyczna, jak głoszą klechdy założycielskie państwa polskiego. Podała FDA do sądu i wygrała apelację.
Reklama
Amerykański sąd apelacyjny stanął po stronie Vanda Pharmaceuticals i uchylił decyzję FDA, która odmówiła rozszerzonego stosowania Hetlioz (tasimelteon) w leczeniu zespołu jet lag bez przeprowadzenia rozprawy.
W orzeczeniu sąd stwierdził, że decyzja FDA o odmowie przesłuchania przedstawicieli Vandy była arbitralna i kapryśna.
Ogłaszając decyzję w poniedziałek sąd apelacyjny zaznaczył, że Vanda wyraźnie przedstawiła znaczące dowody na skuteczność tasimelteonu w łagodzeniu zaburzeń snu i, że każde z jej badań wykazało statystycznie istotną poprawę mierzonego pierwszorzędowego punktu końcowego.
Reklama
Sąd dodał, że odpowiedzi na konkretne, uzasadnione i zakorzenione w dowodach opinie ekspertów, które Vanda Pharmaceuticals przedstawiła w trakcie postępowania dopuszczającego przed FDA, agencja potraktowała pobieżnie.
W związku z tym zdecydowano o przekazaniu sprawy z powrotem do rozpoznania przez FDA.
Działo się w Polsce
Wtorek (19.8.2025)
- Medinice podało, że zakończyło badanie kliniczne wyrobu medycznego PacePress (system przeznaczony do ograniczenia powikłań po implantacji urządzeń kardiologicznych CIED), a w efekcie testu otrzymało pozytywną analizę końcową.
Reklama
"Uzyskane dane stanowią solidną podstawę do dalszych działań zmierzających do komercjalizacji technologii PacePress, której celem jest poprawa jakości opieki nad pacjentami oraz zwiększenie efektywności procedur medycznych. W ocenie zarządu zakończenie badania, stanowi istotny krok przybliżający spółkę do rynkowej komercjalizacji projektu" - oceniła spółka.
Badanie zostało przeprowadzone na terenie Polski, w sześciu ośrodkach klinicznych, z udziałem 122 pacjentów. Analiza wyników wykazała przewagę terapii z wykorzystaniem PacePress w zakresie minimalizacji ryzyka powikłań krwotocznych nad grupą uczestników badania, którzy używali opatrunków standardowych. Parametryzacji tej przewagi brak.
Reklama
Piątek (22.8.2025)
- Diagnostyka dokonała dwóch przejęć konkurencyjnych podmiotów na łączną kwotę 10,6 mln zł. W ramach tych transakcji nabyła 100% udziałów w Niepublicznym Zakładzie Opieki Zdrowotnej Pracownia Genetyki Nowotworów sp. z o. o. oraz pakiet większościowy (51%) w VITA-SKAN sp. z o. o.
"Konsekwentnie realizujemy naszą długoterminową strategię rozwoju, której elementami są selektywne przejęcia w obszarze diagnostyki laboratoryjnej i obrazowej oraz konsolidacja kompetencji w kluczowych obszarach genetyki medycznej. Od początku tego roku zrealizowaliśmy 9 akwizycji, w tym 2 w obszarze diagnostyki obrazowej. Od 2023 roku rozwijamy się w tym perspektywicznym segmencie, co ma znaczenie dla dynamiki wzrostu Grupy, a jednocześnie stanowi wypełnienie naszej misji wspierania ludzi w dłuższym i zdrowym życiu poprzez zwiększanie dostępności nowoczesnej diagnostyki obrazowej. Rośniemy również w ważnym dla pacjentów obszarze genetyki medycznej. Dzięki dołączeniu do Grupy Diagnostyka silnego zespołu specjalistów w dziedzinie onkogenetyki wzmocniliśmy nasz potencjał w obszarze profilaktyki onkologicznej, a także poradnictwa genetycznego w chorobach nowotworowych" - powiedział Dariusz Zowczak, wiceprezes zarządu spółki.
- Rząd zamierza przyjąć w IV kw. br. projekt nowelizacji prawa farmaceutycznego, którego celem jest uchylenie całkowitego zakazu reklamy aptek ogólnodostępnych i punktów aptecznych. Temat jest pokłosiem skargi złożonej w 2013 r. do Komisji Europejskiej (KE). KE uznała skargę za zasadną i skierowała sprawę do Trybunału Sprawiedliwości Unii Europejskiej (TSUE). TSUE 19 czerwca 2025 r. orzekł, że całkowity zakaz jest środkiem nadmiernie restrykcyjnym i nieproporcjonalnym w stosunku do celu ochrony zdrowia publicznego. Za przygotowanie projektu regulacji odpowiedzialny jest resort zdrowia.
Działo się na świecie
Poniedziałek (18.8.2025)
- Reunion Neuroscience poinformował, że jego psychodeliczny kandydat na lek podobny do psylocybiny RE104, testowany w II fazie badań klinicznych znacznie obniżył objawy choroby u kobiet z umiarkowaną do ciężkiej depresji poporodowej (PPD).
"Jesteśmy zachęceni wynikami naszego badania fazy II RECONNECT, które zapewniają silną walidację kliniczną RE104 jako dobrze tolerowanego i skutecznego leczenia PPD, oferującego szybką ulgę przy minimalnych przerwach w codziennych czynnościach" – powiedział Mark Pollack, dyrektor medyczny Reunion.
W badaniu RECONNECT, sfinansowanym za pomocą zeszłorocznej emisji akcji w kwocie 103 mln dol., wzięło udział 84 kobiety w wieku od 21 do 45 lat. Dostawały one podskórnie jednorazową dawkę 30 mg RE104 lub dawkę 1,5 mg psychodeliku w grupie kontrolnej.
Firma podała, że niektórzy pacjenci doświadczyli klinicznie znaczącego zmniejszenia wyniku w skali MADRS w pierwszym dniu po podaniu dawki i utrzymali tę odpowiedź do 28. dnia (w 7. dniu doszło do spadku o 17,2 pkt w skali MADRS), co pozwoliło osiągnąć pierwszorzędowy punkt końcowy badania.
Po tygodniu 77,1% pacjentów, którzy otrzymali wysoką dawkę RE104, miało co najmniej 50% poprawę wyniku MADRS w stosunku do wartości wyjściowej, przy czym 71,4% spełniało kryteria remisji. W grupie kontrolnej te wyniki wyniosły odpowiednio: 61,6% i 41%.
Badany kandydat na lek był ogólnie dobrze tolerowany, a najczęstszymi zdarzeniami niepożądanymi związanymi z leczeniem były nudności (43,9%) i ból głowy (34,1%), które występowały głównie w dniu rozpoczęcia leczenia. Pacjentki nie zgłosiły żadnych poważnych niepożądanych zdarzeń (myśli samobójcze lub zachowania samobójcze).
Przewagą terapii za pomocą RE104 zdaje się to, że 4 godziny po przyjęciu leku, 92,7% pacjentów było gotowych do wypisu, co odróżnia cząsteczkę od innych terapii psychodelicznych, takich jak psylocybina lub LSD, które są związane z dłuższymi doświadczeniami psychoaktywnymi.
Spółka zachęcona odczytem z II fazy klinicznej chce przejść w następnym roku do III fazy. Przed tym krokiem RE104 ma być testowany w badaniu II fazy REKINDLE u pacjentów z zaburzeniami adaptacyjnymi związanymi z rakiem i innymi chorobami medycznymi.
- GoodRx nawiązał współpracę z Novo Nordisk, w ramach której będzie sprzedawać semaglutyd, pod marką Ozempic dla terapii leczenia cukrzycy typu 2 i Wegovy w leczenia otyłości, pacjentom płacącym z własnej kieszeni za 499 dol. miesięcznie, co stanowi około połowę ich obecnej ceny katalogowej.
Good Rx dysponuje siecią ponad 70 tys. aptek w całych Stanach Zjednoczonych. Firma oszacowała, że ok. 19 mln Amerykanów nie ma ubezpieczenia na leki GLP-1, co oznacza, iż nie mają co liczyć na refundację.
W reakcji na wiadomość kurs akcji GoodRx zwyżkował w poniedziałek o 37,3%.
Wtorek (19.8.2025)
- Australijski CSL przeprowadzi restrukturyzację swojego biznesu, której zadaniem jest uproszczenie działalności i zwiększenie wydajności. W tym celu wydzieli do odrębnego podmiotu swoją jednostkę zajmującą się szczepionkami i zmniejszy zatrudnienie nawet o 15%, co ma doprowadzić do rocznych oszczędności w wysokości ponad 500 mln dol.
W związku z redukcją zatrudnienia CSL zapowiedział, że ograniczy swoją działalność badawczo-rozwojową – zmniejszy się liczba placówek tego typu z 11 do 6. Ponadto, w tym miesiącu spółka zamknęła już 22 nierentowne centra plazmowe w USA.
Oszczędności wynikające z tych działań mają zostać osiągnięte w ciągu najbliższych 3 lat, lecz większość z nich zostanie osiągnięta do końca roku podatkowego 2027.
Negatywnie ocenili wieści koncernu inwestorzy, gdyż we wtorek w notowaniach na giełdzie w Australii akcje CSL straciły prawie 17%.
- Komisja Europejska przyznała warunkowe dopuszczenie do obrotu Rezdiffrze (resmetirom) Madrigal Pharmaceuticals. Dzięki temu na terenie UE pojawił się pierwszy lek do terapii stłuszczeniowego zapalenia wątroby związanego z dysfunkcją metaboliczną (MASH).
W planach jest to, że lek najpierw trafi na półki sklepowe w Niemczech w przyszłym kwartale, a dopiero wtedy w ślady naszego zachodniego sąsiada pójdą inne kraje.
"Ta zgoda oznacza historyczny przełom dla pacjentów w Europie żyjących z MASH bez konieczności wykonywania biopsji, aby zakwalifikować się do leczenia i wyraźne skupienie się na populacji o wysokich niezaspokojonych potrzebach" - wyjaśnił Bill Sibold, dyrektor generalny Madrigal Pharmaceuticals.
Zatwierdzenie w Europie nastąpiło rok po dopuszczeniu preparatu do sprzedaży w USA. Ale także w tydzień po tym, gdy FDA rozszerzyła etykietę wskazaniową dla Wegovy Novo Nordisk na terapię MASH. Novo złożyło wniosek o wydanie identycznej decyzji przez europejskiego regulatora w lutym br.
Madrigal czuje oddech konkurencji na plecach w obszarze MASH. Żeby temu przeciwdziałać spółka podpisała w lipcu umowę licencyjną obowiązującą na całym świecie z CSPS Pharmaceutical o maksymalnej wartości 2,1 mld dol. (120 mln dol. z góry, a reszta w miarę osiągania kamieni milowych rozwoju doustnego agonisty receptora GLP-1 i pochodnej orforglipronu, nazwanej przez CSPC SYH2086). Madrigal chce połączyć SYH2086 w terapii skojarzonej z Rezdiffrą.
- FDA przyznała Altimmune zgodę na przyspieszoną ścieżkę rozwojową (Fast Track) dla kandydata spółki na lek – pemwidutide, który ma służyć w terapii zaburzeń związanych z używaniem alkoholu (AUD).
Altimmune prowadzi obecnie rekrutację do badania II fazy klinicznej pod nazwą RECLAIM. Wziąć udział w nim ma około 100 pacjentów podzielonych na dwie grupy: otrzymującą dawkę 2,4 mg pemwidutide lub placebo raz w tygodniu przez okres 24 tygodni.
Pierwszorzędowym punktem końcowym badania jest zmiana w stosunku do wartości wyjściowej średniej liczby dni intensywnego spożywania alkoholu, przy czym kluczowe drugorzędowe punkty końcowe obejmują odsetek osób, u których osiągnięto dwupoziomowe obniżenie poziomu ryzyka picia według kryteriów WHO oraz bezwzględną zmianę w stosunku do wartości wyjściowej średnich poziomów fosfatydyloetanolu (PEth), biomarkera spożycia alkoholu w surowicy.
"Pomimo szacowanego rozpowszechnienia AUD u ponad 28 milionów dorosłych w samych Stanach Zjednoczonych, niedobór skutecznych opcji leczenia stworzył znaczną lukę w leczeniu AUD, przy czym tylko 2% jest obecnie leczonych lekami" - powiedział dr Vipin K. Garg, dyrektor generalny Altimmune.
"Obecnie zatwierdzone terapie wykazały ograniczoną skuteczność i nie są w stanie odpowiednio rozwiązać chorób współistniejących z AUD, takich jak stłuszczenie wątroby, hiperlipidemia i nadciśnienie lub inne choroby współistniejące z otyłością, na które często cierpią osoby z AUD. Oznaczenie Fast Track uwzględnia zarówno pilną, niezaspokojoną potrzebę związaną z AUD, jak i potencjał pemwidutide do odegrania roli w leczeniu tego poważnego schorzenia" - dodał.
Oznaczenie w trybie Fast Track ma na celu ułatwienie opracowywania i przyspieszanie przeglądu terapii, które mogą być stosowane w leczeniu poważnych lub zagrażających życiu schorzeń.
Firmy badające kandydatów na lek z oznaczeniem Fast Track mają możliwość skorzystania z wczesnej i częstej komunikacji z FDA, mogą kwalifikować się do etapowego składania wniosków o dopuszczenie do obrotu i priorytetowego przeglądu wniosku o dopuszczenie do obrotu (NDA).
AUD jest stanem chorobowym spowodowanym upośledzoną zdolnością do zaprzestania lub kontrolowania szkodliwego spożywania alkoholu. Zaburzenia związane z używaniem alkoholu mogą również prowadzić m. in. do chorób wątroby, chorób układu krążenia i raka. Większość pacjentów z zaburzeniami ma współistniejącą nadwagę lub otyłość oraz występuje u nich stłuszczenie wątroby.
WHO oceniło, że szkodliwe spożywanie alkoholu jest siódmą najczęstszą przyczyną zgonów i niepełnosprawności na świecie, a alkohol odpowiada za 50% wszystkich zgonów związanych z wątrobą.
Środa (20.8.2025)
- Jazz Pharmaceuticals nabył licencję globalną od Saniona, która przyznała wyłączne prawa do rozwoju przedklinicznego kandydata na lek na padaczkę SAN2355. Licencjobiorca zapłaci z góry 42,5 mln dol., a dodatkowe 192,5 mln dol. wchodzi w grę w miarę osiągania kolejnych kamieni milowych.
Oprócz tego Saniona kwalifikuje się również do otrzymania do 800 mln dol. w komercyjnych kamieniach milowych oraz wielopoziomowych tantiem, od średnich jednocyfrowych do niskich dwucyfrowych.
Zgodnie z umową Jazz będzie odpowiedzialny za przyszłe działania rozwojowe, regulacyjne i komercjalizacyjne dla małocząsteczkowego aktywatora kanałów potasowych Kv7.2/Kv7.3.
Aktualnie Jazz Pharmaceutical koncentruje się na obszarze onkologii, skąd pochodzi większość jej portfolio produktowego. Mimo to dwa leki generujące największą sprzedaż to preparaty z dziedziny neurologii. Xywav, służący terapii narkolepsji, wygenerował 760 mln dol. sprzedaży w I półroczu 2025, a doustny roztwór kannabidiolu, sprzedawany jako Epidiolex na terenie USA i Epidyolex w Europie zapewnił w tym okresie 470 mln dol.
Czwartek (21.8.2025)
- Dawnzera (donidalorsen) Ionis Pharmaceuticals, kandydat na lek przeciw atakom dziedzicznego obrzęku naczynioruchowego (HAE) u dorosłych i dzieci w wieku co najmniej 12 lat, dostał zielone światło na dopuszczenie od FDA.
Dawnzera została zatwierdzona na podstawie wyników badania III fazy klinicznej OASIS-HAE. Po 24 tygodniach u osób, które otrzymywały raz w miesiącu lek w zastrzyku, zaobserwowano średnie zmniejszenie liczby napadów HAE o 81% w porównaniu z placebo.
Oprócz tego otwarte badanie przedłużające wykazało, że substancja doprowadziła do całkowitego zmniejszenia średniego wskaźnika ataków o 94% w stosunku do wartości wyjściowej po upływie jednego roku.
- Dynavax Technologies podzielił się wynikami badań szczepionki przeciwko półpaścowi, która według sponsora wypadła korzystnie w porównaniu z przebojową szczepionką Shingrix GSK, która przyniosła gigantowi Big Pharmy 1,7 mld funtów przychodów w I półroczu 2025.
Z-1018, szczepionka rozwijana przez Dynavax, łączy wyprodukowany antygen glikoproteiny E (gE) z CpG 1018, zastrzeżonym adiuwantem szczepionkowym spółki. W badaniu jest ona porównywana z Shingriksem.
Spółka ujawniła, że miesiąc po zaaplikowaniu drugiej dawki Z-1018 otrzymano 100% humoralny wskaźnik odpowiedzi na szczepionkę oraz 89,7% komórkową odpowiedź immunologiczną na szczepionkę, w porównaniu z analogicznymi wskaźnikami odpowiednio 96,9% i 93,5% dla Shingriksu.
Ogólnie szczepionka Dynavax miała złożony wskaźnik odpowiedzi na szczepionkę wynoszący 89,7% w porównaniu z 90,3% w przypadku Shingrix.
Podane wyniki przez Dynavax wzbudziły chwilowe zainteresowanie inwestorów, ponieważ w czwartek kurs akcji korporacji poszedł w górę o 5%, lecz dzień później wrócił niemal do punktu wyjścia po 4% korekcie cenowej.
"Te pozytywne dane wyznaczają ważny punkt zwrotny dla naszego nowatorskiego programu szczepień przeciwko półpaścowi, ponieważ staramy się opracować produkt o potencjalnie najlepszym w swojej klasie profilu, którego celem jest zakłócenie wielomiliardowego rynku szczepionek przeciwko półpaścowi, obecnie zdominowanego przez jeden produkt" – wyjawił Ryan Spencer, dyrektor generalny Dynavax.
"Osiągnęliśmy nasz cel w tym badaniu, ponieważ wyniki pokazują odpowiedź immunologiczną porównywalną z Shingrix, wraz z korzystnym profilem tolerancji i stanowią podstawę do wyboru dawki i schematu, aby przejść do dalszego rozwoju" - dodał Spencer.
Piątek (22.8.2025)
- AbbVie planuje złożenie wniosków o rejestrację leku Rinvoq (upadacitinib) w leczeniu wypadania włosów po tym, gdy uzyskała korzystne wyniki w dwóch badaniach III fazy klinicznej dla inhibitora JAK w ramach programu klinicznego UP-AA.
W dwóch badaniach głównych wzięło udział 1 399 pacjentów w wieku od 12 do 64 lat z ciężkim łysieniem plackowatym i średnim wyjściowym pokryciem włosów na skórze głowy wynoszącym około 16%. Uczestnicy zostali losowo przydzieleni do grupy przyjmującej doustnie raz na dobę dawkę 15 mg lub 30 mg Rinvoq albo placebo.
W badaniu pierwszym 55% pacjentów, którzy otrzymali większą dawkę Rinvoq, i 45,2% pacjentów, którzy przyjmowali mniejszą dawkę, doświadczyło pokrycia włosów na skórze głowy na poziomie 80% lub więcej w 24. tygodniu, co odpowiada nasileniu łysienia (SALT) w skali ≤ 20. W grupie placebo analogiczny odczyt wyniósł 1,5%.
Wyniki te były zgodne z badaniem drugim, w którym 54,3% i 44,6% pacjentów przyjmujących odpowiednio 30 mg i 15 mg Rinvoq osiągnęło 80% lub więcej pokrycia włosów na skórze głowy w 24. tygodniu.
Obserwuj nas na  Google News
Google News
Chcesz być na bieżąco z wieściami z naszego portalu? Obserwuj nas na Google News!
Źródło i opracowanie własne
Aktualizacja: 26/08/2025 11:30

Reklama
Komentarze opinie
Podziel się swoją opinią
Twoje zdanie jest ważne jednak nie może ranić innych osób lub grup.





Reklama
Reklama




Reklama
Reklama
Reklama
Najnowsze wiadomości
- 18/07 Telemedycyna dla seniorów: co oferuje NFZ?
- 18/07 Wyrabiamy kartę EKUZ bez wychodzenia z domu. Zajmuje to tylko kilka minut
- 18/07 AOTMiT o otyłości. Kompleksowa opieka bariatryczna bliżej koszyka świadczeń
- 17/07 Symfonia życia w Kajetanach. Co muzyka robi z mózgiem osoby z implantem ślimakowym?
- 17/07 Kriomedpol zmienia zarząd. Nowe otwarcie dla producenta urządzeń do krioterapii i kriochirurgii
- 17/07 Prawie 16 tys. kandydatów na WUM. Uczelnia: „Jesteśmy liderem pod względem bezwzględnej liczby kandydatów”
- 17/07 NIK: lekarz pracował 144 godziny bez przerwy. Kontrola SOR-ów ujawniła przeciążenie personelu i systemowe braki
- 17/07 „Setki milionów złotych miesięcznie zagrożone”. PAŻP walczy o odblokowanie środków, jest formalny sprzeciw do sądu w Brukseli
- 17/07 Kadry. Ppłk Agata Będzichowska została Komendantem CSK MON WIM-PIB
- 17/07 Jest podpis prezydenta. Kiedy wystartuje Bon Senioralny?
- 17/07 Dietetycy dostaną własną ustawę. Wioleta Tomczak: Zachęcam do konsultacji publicznych
- 17/07 Kotula po wecie ustawy o statusie osoby najbliższej: „To nie blokada, a opóźnienie zmiany prawa”
- 17/07 Sześć nowych projektów dla ochrony zdrowia. „Obowiązek przepracowania przynajmniej pół etatu w jednym szpitalu”
- 17/07 Prezydent zawetował ustawę o statusie osoby najbliższej. „Nie podpiszę alternatywy dla małżeństwa”. Podpisane dwie inne ustawy
- 17/07 Pokonali raka, ale stracili głos. Onkologopeda: rehabilitacja powinna zacząć się przed leczeniem
- 17/07 Mniej niż 20 proc. dzieci jest wystarczająco aktywnych. Dr Marta Łabęcka: kondycję fizyczną przedszkolaków trzeba diagnozować wcześniej
- 16/07 „Bez dobrej diety nie będzie progresu”. Dietetyk: wyniki sportowe zaczynają się w kuchni
- 16/07 Taka ankieta przed rezonansem to za mało. Rzecznik Praw Pacjenta upomina
- 16/07 Ochłodzenie skóry pomogło pacjentkom zachować włosy podczas chemioterapii
- 16/07 E-skierowanie: jak działa i jak korzystać?
Komentarze