
Reklama
W świecie fantazji zdają się tkwić menedżerowie ImmunityBio, gdyż po raz trzeci w ostatnich miesiącach firma dostała od FDA pismo ostrzegawcze dotyczące kampanii reklamowej. Regulator uważa, że firma solidnie przesadziła w przekazie komunikacyjnym, w którym zachwala Anktivę, preparat zatwierdzony dla leczenia raka pęcherza, jako zdolną do terapii wszystkich nowotworów i działania jak szczepionka przeciw rakowi. Merck wyda 6,7 mld dol. na przejęcie Terns Pharma, która rozwija TERN-701, potencjalnego blockbustera w terapii przewlekłej białaczki szpikowej. Mniej głęboko do kieszeni sięgnęli inni przedstawiciele Big Pharmy w transakcjach Gilead Sciences/Ouro Medicines i Novartis/Excellergy. Kodiak Sciences ogłosił, że Zenkuda (tarcocimab tedromer) testowana w badaniu III fazy klinicznej GLOW2 osiągnęła główny cel u pacjentów z uszkodzeniem wzroku związanym z cukrzycą - 62,5% pacjentów leczonych preparatem osiągnęło co najmniej dwustopniową poprawę w skali nasilenia retinopatii cukrzycowej. Najmłodsi dostali nowe narzędzie do leczenia ciężkiego niedoboru adhezji leukocytów typu I (LAD-I), gdyż FDA dopuściła do obrotu Kresladi (marnetegragene autotemcel). Akcje Wave Life Sciences przepołowiły się jednego dnia po podaniu odczytu danych z I fazy klinicznej z badania INLIGHT dla WVE-007, rozwijanego pod kątem leczenia otyłości. Część analityków jest zdania, że reakcja była przesadzona, a perspektywy dla leku są o wiele lepsze niż uważa rynek giełdowy.
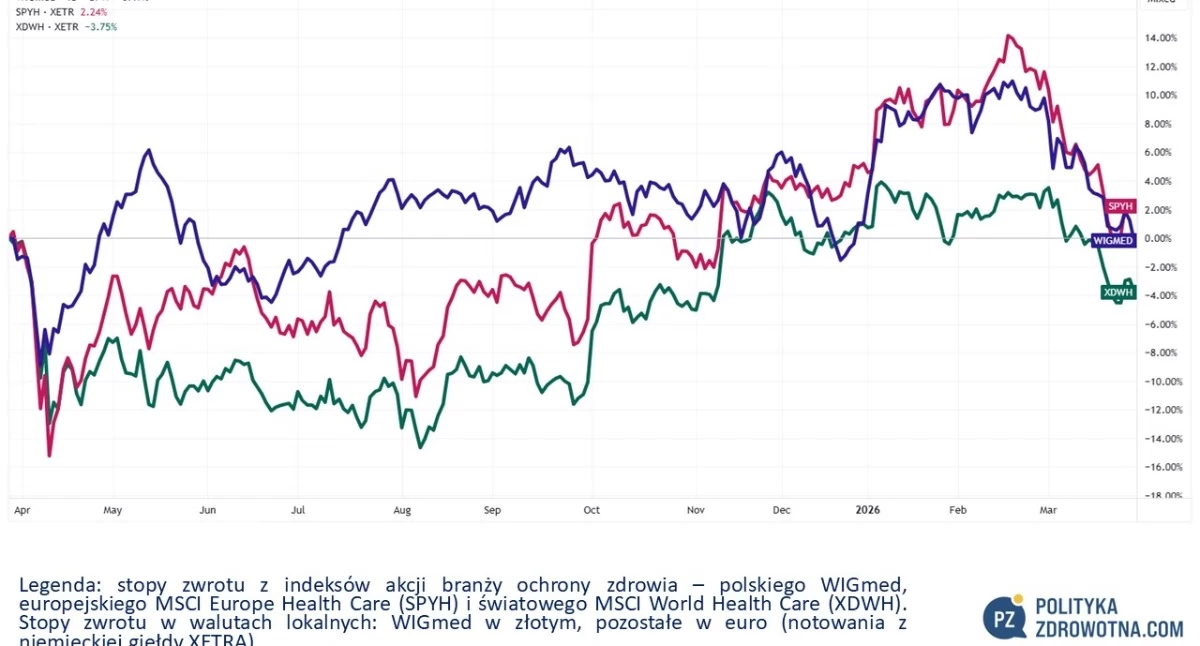
WIGmed, polski indeks akcji spółek ochrony zdrowia, zniżkował w tym tygodniu giełdowym 0,8%. Lepiej działo się na parkietach zagranicznych, gdyż MSCI Europe Health Care (SPYH) poszedł w górę 0,9%, zaś globalny MSCI World Health Care (XDWH) zyskał 0,2%.
Rynki finansowe wciąż pozostawały pod wrażeniem wojny w Zatoce Perskiej pomiędzy koalicją USA i Izraela a Iranem, której wybił właśnie miesiąc od wybuchu. W przestrzeni publicznej sporo mówi się o zakłóceniach łańcuchów dostaw w związku z blokadą Cieśniny Ormuz. Niewiele w tych doniesieniach z kolei o wpływie konfliktu na wyniki spółek biofarmaceutycznych.
Reklama
Analitycy firmy inwestycyjnej Truist Securities uważają, że bezpośrednie oddziaływanie wojny w Zatoce Perskiej, w którą Iran uwikłał również państwa Półwyspu Arabskiego, stroniące od ataków kinetycznych, nie wpłynie istotnie na wyniki finansowe branży - Bliski Wschód w niewielkim stopniu przyczynia się bowiem do całokształtu sprzedaży koncernów. Według Truist Securities największą ekspozycję na region mają GSK i Takeda, które czerpią 6% sprzedaży z Bliskiego Wschodu.
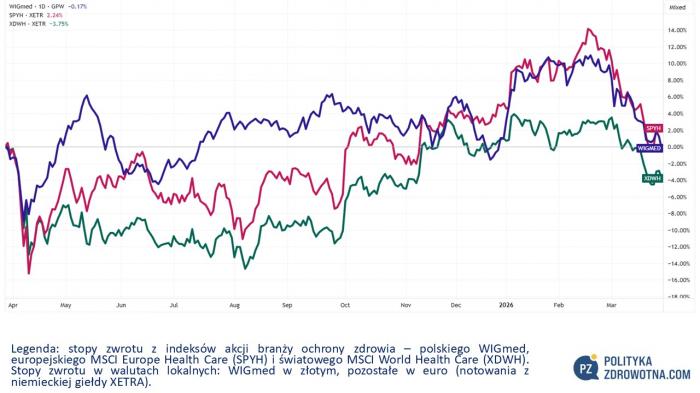
Źródło: TradingView
W Top20 MSCI World Health Care najbardziej wyróżniły się papiery Merck (+4,8%), AstraZeneca (+2,4%) oraz ex-aequo Johnson & Johnson i AbbVie (+2,1%). Najbardziej niepocieszeni okazali się właściciele walorów, które najwięcej straciły - UnitedHealth Group (-6,1%), Intuitive Surgical (-5,3%) i Vertex Pharmaceuticals (-4,6%).
Reklama
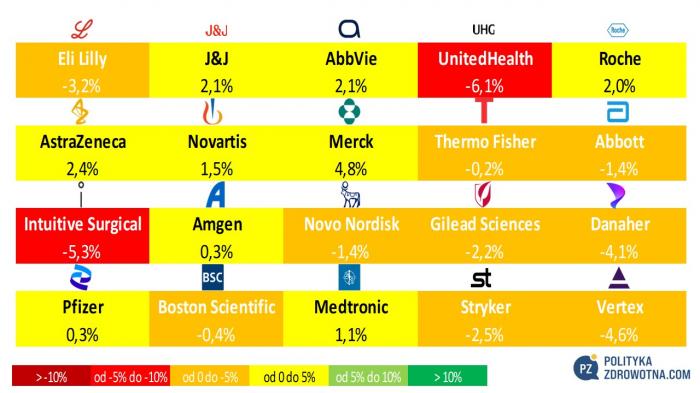
Źródło: opracowanie własne na podstawie stooq.pl
W Polsce gwiazdą tygodnia bezsprzecznie było Medinice (+40,8%). W czołowej trójce najlepiej zachowujących się akcji uplasowały się również Captor Therapeutics (+2,6%) i Mabion (+2,1%). Największe spadki zanotowały PolTREG (-30,4%), Molecure (-15,3%) i Enel-Med (-11,1%).
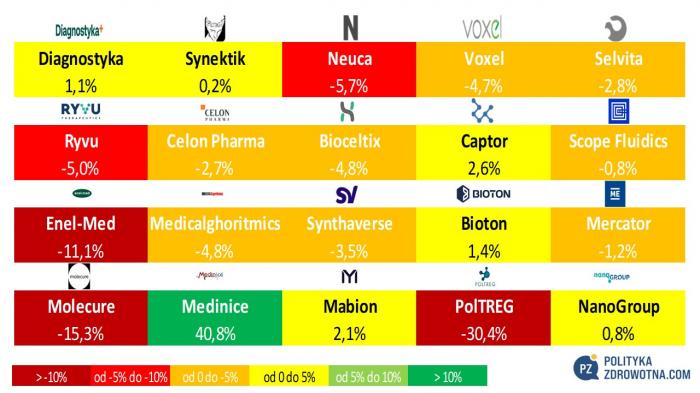
Źródło: opracowanie własne na podstawie stooq.pl
Działo się w Polsce
Wtorek (24.3.2026)
- Pure Biologics ocenił, że wyniki badania fazy 0 kandydata na lek PBA-0111 potwierdzają projektowany mechanizm działania u ludzi i wspierają zasadność dalszego rozwoju klinicznego.
Reklama
"Badanie kliniczne (faza 0) kandydata na lek PBA-0111 przeprowadzono w USA i miało charakter wieloośrodkowy i otwarty. Zrekrutowanych zostało 6 pacjentów z mięsakiem tkanek miękkich. Celem badania było potwierdzenie mechanizmu działania cząsteczki u ludzi na poziomie molekularnym i komórkowym. Pacjentom podano mikrodawki PBA-0111 bezpośrednio do guza, a następnie - w czasie od 2 do 96 godzin - usunięto guzy i poddano je analizie biomarkerów - molekularnych oznak potwierdzających działanie kandydata na lek zgodnego z założonym mechanizmem. Badania potwierdziły obecność celu molekularnego, GARP na poziomie mRNA oraz komórek układu odpornościowego w mikrośrodowisku guza" – podał zarząd spółki.
Firma przyznała, że nie zaobserwowano jednoznacznych dowodów na bezpośrednie zabijanie komórek nowotworowych w miejscu podania leku, co jej zdaniem może jednak wynikać z niskiego poziomu białka GARP, ograniczonej liczby komórek efektorowych lub niewystarczającego czasu ekspozycji guza na PBA-0111 w warunkach badania fazy 0.
Reklama
U 5 z 6 pacjentów zaobserwowano oznaki działania immunomodulacyjnego. U 2 z 6 pacjentów dostrzeżono zmniejszenie liczby immunosupresyjnych limfocytów T regulatorowych (Treg), których zabijanie jest jednym z głównych mechanizmów działania PBA-0111. U 5 z 6 pacjentów wykazano tendencję do wzrostu aktywności cytotoksycznych limfocytów T, mierzoną poziomem granzymu B, co może świadczyć o aktywacji odpowiedzi immunologicznej w guzie na skutek zmniejszenia immunosupresji ze strony środowiska guza.
Zebrane dane sugerują, że w guzach z niskim poziomem GARP, PBA-0111 działa przede wszystkim poprzez hamowanie szlaku TGF-β1, albo na skutek blokowania kompleksu GARP-TGF-β1, albo zabijanie limfocytów Treg. Mechanizm ten prowadzi do zmniejszenia immunosupresji w guzie i aktywacji komórek efektorowych układu odpornościowego, co może wzmacniać immunologiczną odpowiedź przeciwnowotworową.
Reklama
"Wyniki są spójne z wcześniejszymi badaniami przedklinicznymi w modelach mysich, w których obserwowano aktywację limfocytów T CD8+ oraz zahamowanie wzrostu guzów. Zarząd spółki ocenia, że uzyskane wyniki potwierdzają zamierzony mechanizm działania PBA-0111 u ludzi oraz wspierają zasadność dalszego rozwoju klinicznego projektu. Jednocześnie wskazują na konieczność stratyfikacji pacjentów pod kątem obecności białka GARP" – ocenił Pure Biologics.
Na wieść o odczycie badania kurs akcji Pure Biologics od wtorku przez trzy kolejne sesje rósł w tempie zbliżonym do 40% dziennie, po czym skorygował się o blisko 20% w piątek, co doprowadziło do tygodniowej zwyżki papierów o ok. 110%.
Reklama
- Pharmena zakończyła proces opracowania i walidacji metod oznaczeń zawartości substancji czynnych w przyszłym wyrobie medycznym do stosowania w trudno gojących się ranach.
"Prace nad naszym wyrobem medycznym weszły w kluczową fazę, w której każdy z końcowych etapów ma fundamentalne znaczenie dla osiągnięcia oczekiwanego efektu jakościowego i terapeutycznego. Zakończenie walidacji metod analitycznych to nie tylko istotny kamień milowy projektu, ale także potwierdzenie, że rozwijamy produkt w oparciu o najwyższe standardy jakości i bezpieczeństwa, zgodne z GMP. Wierzymy, że innowacyjna formuła naszego rozwiązania, przeznaczonego do stosowania również w warunkach domowych, może realnie zmienić podejście do leczenia trudno gojących się ran - zwiększając dostępność terapii, skracając czas leczenia i ograniczając ryzyko powikłań. Naszą ambicją jest wprowadzenie na rynek produktu, który stanie się nowym standardem postępowania i przyniesie pacjentom wymierne korzyści" - powiedziała Marzena Wieczorkowska, wiceprezes zarządu Pharmeny.
Reklama
Środa (25.3.2026)
- Molecure pozyskał ok. 21 mln zł brutto z emisji 4 120 531 akcji serii K. Pozyskany kapitał posłuży do kontynuacji kluczowych badań klinicznych - fazy II badania KITE (OATD-01) oraz fazy I badania w onkologii (OATD-02) - a także wesprze rozwój platformy AI.
"Zbliżamy się do etapu, w którym oba nasze programy kliniczne powinny dostarczyć istotnych danych, otwierając przed Molecure kolejny etap budowy wartości. Osiągnięcie ważnego kamienia milowego w badaniu KITE (OATD-01), czyli zrandomizowanie pierwszych 30 pacjentów, pozwala nam z optymizmem oczekiwać na wyniki pierwszej analizy cząstkowej już w połowie 2026 roku. Równolegle w programie OATD-02, w badaniu fazy pierwszej mamy pozytywne odczyty parametrów bezpieczeństwa, a co kluczowe - zaobserwowaliśmy także zamierzony efekt farmakodynamiczny, czyli kliniczne potwierdzenie mechanizmu działania leku u pacjentów onkologicznych. To znacząco przybliża nas do celu wyznaczenia dawki aktywnej farmakologicznie w najbliższych miesiącach" – ocenił Marcin Szumowski, prezes zarządu Molecure.
Reklama
"Równolegle rozwijamy platformę opartą o narzędzia AI wspierająca proces odkrywania leków poprzez zwiększenie efektywności wyłaniania kandydatów o dobrych właściwościach lekopodobnych dla zupełnie nowych celów terapeutycznych. Widzimy również potencjał jej wykorzystania w modelu współpracy z partnerami zewnętrznymi. Projekt ten jest współfinansowany grantem w wysokości 27 mln zł" - dodał.
Piątek (27.3.2026)
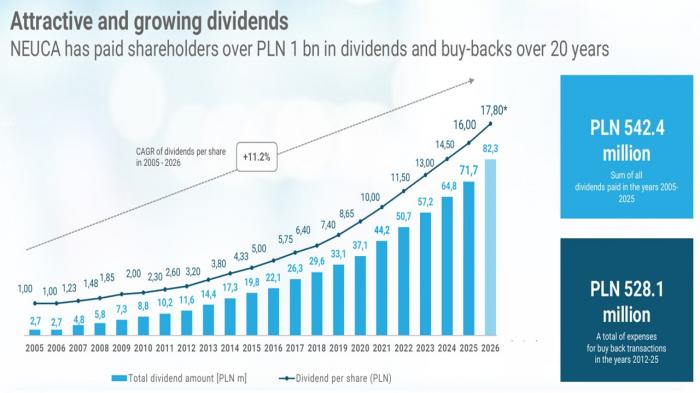
- Neuca spodziewa się w tym roku poprawy rentowności całego segmentu badań klinicznych oraz oczekuje, że sfinalizuje akwizycję w obszarze SMO na nowym rynku. Spółka planuje również wypłacić rekordową w swojej historii dywidendę na akcję - w kwocie 17,8 zł.
Reklama
"W 2026 roku spółka będzie koncentrować się na komercjalizacji strategii zintegrowanego modelu serwisów onkologicznych, w tym na unifikacji strategii sprzedażowej oraz dalszym rozwoju platformy P1. Równolegle, w segmencie SMO, kontynuowany będzie organiczny rozwój nowych ośrodków onkologicznych i neurologicznych. Jednocześnie oczekujemy zauważalnego wzrostu biznesu onkologii oraz nowego biznesu w tym obszarze - zarówno w Europie, jak i w USA - na poziomie wszystkich segmentów (przychód i NNB)" – wskazała Neuca.
Spółka odnotowała w segmencie badań klinicznych w 2025 r. stratę operacyjną w wysokości 9,9 mln zł - rok wcześniej było 16 mln zł zysku.
Reklama
W IV kw. 2025 r. toruńska firma dokonała akwizycji sieci ośrodków w USA - P1 Trials, specjalizujących się w onkologicznych badaniach wczesnofazowych.
"Platforma P1 skupia się na wczesnofazowych badaniach klinicznych w onkologii. Pokrywają się one w całości z obszarem kompetencji zarówno Ośrodków Onkologicznych Pratia w Europie, jak i naszego CRO Kapadi. W ramach brandu P1 będziemy konsolidować cały obszar SMO wczesnofazowych badań klinicznych w onkologii. Po transakcji udało się nam rozpocząć pierwsze projekty kliniczne, które w pierwszym kwartale 2026 r. już generują przychody. Obecnie mamy kilkanaście projektów wczesnofazowych w etapie preselekcji i spodziewamy się rozpoczęcia kilku, do kilkunastu badań jeszcze w tym roku" – wyjaśniła Neuca.
"Jeśli chodzi o kolejne akwizycje, to spodziewamy się zamknięcia jeszcze jednej transakcji w tym roku. Nie wykluczamy jednak bardziej kreatywnych modeli typu JV w ramach strategicznych inicjatyw w onkologii" - dorzuciła.
Spółka oczekuje wzrostu wartości rynku hurtu aptecznego w 2026 r. w wysokości ok. 6,0-6,5%.
"W sezonie infekcyjnym 2025/2026 liczba zachorowań nie odbiegała istotnie od stosunkowo wysokiego poziomu z ubiegłego roku. Obserwowaliśmy natomiast przesunięcie szczytu zachorowań w czasie. Ponadto w związku z bardzo dużą liczbą szczepień przed sezonem grypowym w roku ubiegłym widzimy, iż dotkliwość zachorowań istotnie spadła, co przełożyło się również na zmniejszony wolumen leków przeciwgrypowych i pochodnych" – zaznaczyły władze Neuki.
"Średni wzrost rynku hurtowej dystrybucji leków w I kwartale bieżącego roku wydaje się oscylować w okolicach 3%, z prognozowanym wzrostem w marcu w przedziale 5-6%. Jest to poniżej naszych wstępnych oczekiwań średniorocznego wzrostu rynku na poziomie 6 pkt proc., na ten moment jest jednak zbyt wcześnie na rewizję naszej pierwotnej prognozy" - dodały.
Źródło: Neuca
- PolTREG podał, że podczas procesu przyspieszonego badania księgi popytu inwestorzy złożyli deklarację objęcia 851 052 akcji nowej emisji oraz nabycia 401 956 akcji istniejących spółki po cenie 15 zł, co daje łącznie ok. 18,8 mln zł. Zamiarem spółki było pozyskanie w ramach inicjatywy ok. 30 mln zł.
Pieniądze zostaną przeznaczone na projekty o najwyższym potencjale terapeutycznym i komercyjnym, w tym doprowadzenie projektu CAR-Treg (komórkowa terapia m.in. stwardnienia rozsianego i stwardnienia zanikowego bocznego) do etapu rozpoczęcia badania klinicznego fazy I, co PolTREG planuje zapoczątkować w II połowie br. Poza tym środki z transakcji mają sfinansować rozwój terapii in vivo (tzw. "szczepionki mRNA na cukrzycę"), która ma osiągnąć etap Proof of Concept (PoC) w badaniach na zwierzętach w I połowie 2027 r.
Działo się na świecie
Poniedziałek (23.3.2026)
- Pfizer i Valneva poinformowały, że choć ich badana szczepionka przeciw boreliozie (LB6V) w III fazie klinicznej osiągnęła skuteczność 73,2%, to nieznacznie nie spełniła głównego punktu końcowego w postaci dolnego przedziału ufności statystycznej.
LB6V zdaje się najbardziej zaawansowaną szczepionką przeciw boreliozie w fazie rozwoju i celuje w sześć serotypów bakterii Borrelia burgdorferi wywołującej tę odkleszczową chorobę. Substancja była testowana przez konsorcjum amerykańsko-francuskich naukowców podczas badania VALOR, które objęło 9 437 uczestników w wieku co najmniej 5 lat. Partnerzy badawczy, którzy rozwijają szczepionkę od kwietnia 2020 r. zamierzają przystąpić do zgłoszeń regulacyjnych do FDA i EMA jeszcze w tym roku.
"Biorąc pod uwagę klinicznie istotną skuteczność oraz fakt, że dolna granica 95% przedziału ufności była powyżej 20 w drugiej wcześniej określonej analizie, jesteśmy przekonani o potencjale szczepionki i planujemy złożenie wniosków do organów regulacyjnych" - podał amerykański koncern Big Pharmy.
Oznaki rozczarowania wynikami badania wykazali analitycy kanadyjskiego banku inwestycyjnego RBC, którzy przyznali jednak, że gdy wniosek o zatwierdzenie LB6V trafi do FDA, to "może spotkać się z wyrozumiałym uchem", ponieważ amerykański sekretarz zdrowia i opieki społecznej Robert F. Kennedy Jr. nie raz podkreślał potrzebę rozwiązania problemu boreliozy. W tym celu powołał do życia w grudniu 2025 panel pacjentów z boreliozą oraz specjalistów z tej dziedziny, aby wzmocnić profilaktykę, diagnozę i długoterminową opiekę nad osobami dotkniętymi tą chorobą. Wagę sprawy wzmacnia to, że obecnie nie ma dostępnej szczepionki przeciwko boreliozie.
Walka z boreliozą ma już historyczny precedens, gdy w grudniu 1998 r. GSK dostało zatwierdzenie dla szczepionki LYMERix o podobnej skuteczności (75% po roku od zaszczepienia). Mimo oferowania jej przez kilka lat GSK wycofało produkt ze sprzedaży z powodu słabego popytu i obaw społecznych o działania niepożądane.
RBC oceniło, że szczyt sprzedaży LB6V sięgnie 525 mln dol., o ile szczepionka dostanie zielone światło od FDA.
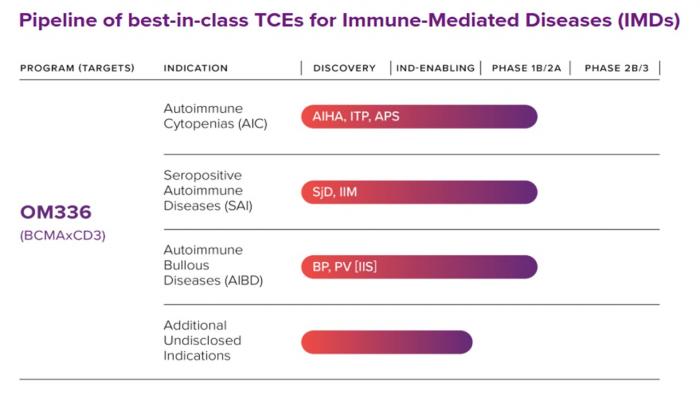
- Gilead Sciences wyda do ok. 2,2 mld dol. na prywatną firmę biotechnologiczną Ouro Medicines, skupionej na opracowywaniu terapii stymulujących limfocyty T w chorobach autoimmunologicznych. Dzięki transakcji do portfela w dziedzinie chorób zapalnych Gilead Sciences trafi OM336 (gamgertamig), cząsteczka rozwijana m. in. pod kątem cytopenii autoimmunologicznych (np. niedokrwistości hemolitycznej i trombocytopenii immunologicznej).
Gamgertamig otrzymał od USA certyfikaty Fast Track oraz Orphan Drug Designation (Fast Track) oraz oznaczenie leku sierocego (Orphan Drug) w leczeniu anemii i trombocytopenii i oczekuje się, że rozpocznie badania rejestracyjne w 2027 r.
Gilead zapłaci 1,675 mld dol. z góry gotówką, a ponadto 500 mln dol. dodatkowo po osiągnięciu określonych kamieni milowych. W konfiguracji biznesowej Gilead/Ouro jest zdaje się też miejsce dla Galapagos z siedzibą w belgijskim Mechelen. Gilead Sciences ujawnił, że prowadzi obecnie zaawansowane rozmowy z Galapagos dotyczące potencjalnej współpracy badawczo-rozwojowej nad przejętymi aktywami Ouro Medicines.
Układ między Gileadem a Galapagos ma obejmować m. in. następujące kluczowe warunki:
- Galapagos zapłaci 50% wkładu z góry oraz 50% wszelkich warunkowych płatności milowych należnych akcjonariuszom Ouro Medicines,
- Galapagos przejęłoby w dużej mierze wszystkie aktywa operacyjne Ouro Medicines i zatrzymało pracowników,
- Gilead i Galapagos miałyby współpracować przy rozwoju OM336, przy czym Galapagos odpowiadałoby za koszty rozwoju poprzez rozpoczęcie badań rejestracyjnych, a koszty rejestracji studiów byłyby podzielone równo między strony,
- Gilead zachowałby wyłączne światowe prawa do komercjalizacji (z wyjątkiem Wielkich Chin, gdzie Keymed Biosciences posiada istniejące prawa do komercjalizacji), a Gilead płaciłby Galapagosowi tantiemy w wysokości 20%-23% od sprzedaży netto.
Źródło: Ouro Medicines
- Kali Therapeutics zawarła umowę licencyjną z Sanofi w celu opracowania eksperymentalnej terapii przeznaczonej do leczenia kilku chorób autoimmunologicznych. Francuzi zapłacą z góry 180 mln dol., a Kali jest uprawnione do dodatkowych 1,05 mld dol. w zależności od spełnienia kamieni milowych rozwojowych i komercyjnych. Jeśli terapia zostanie skomercjalizowana licencjodawca może dostać stopniowo rosnące tantiemy sprzedażowe w przedziale od wysokich jednocyfrowych do dwucyfrowych wartości procentowych.
Sanofi otrzymało wyłączne globalne prawa do KT501, nowego trójspecyficznego przeciwciała opracowanego na platformie badawczej Kali. Cząsteczka została zaprojektowana tak, aby regulować komórki odpornościowe wywołujące choroby autoimmunologiczne, takie jak toczeń i reumatoidalne zapalenie stawów. KT501 znajduje się obecnie w badaniu klinicznym wczesnej fazy (I), której celem jest ocena bezpieczeństwa i tolerancji u pacjentów z reumatoidalnym zapaleniem stawów.
- Cencora, spółka specjalizujące się w dystrybucji leków, zapowiedziała, że za 1,1 mld dol. kupi biznes siatkówkowy EyeSouth Partners, wskutek czego powiększy swoje kompetencje w zakresie opieki okulistycznej. Po sfinalizowaniu transakcji lekarze okuliści specjalizujący się w siatkówce EyeSouth Partners przejdą do Retina Consultants of America należącej do Cencory, czołowej organizacji świadczącej usługi zarządzania medycyną w tej dziedzinie.
Wtorek (24.3.2026)
- ImmunityBio otrzymała od FDA pismo ostrzegawcze, w którym nadzorca stwierdził, że reklama telewizyjna i podcasty promujące terapię onkologiczną spółki były fałszywe lub wprowadzały w błąd, naruszając prawo federalne. Agencja uznała, że przekaz komunikacyjny, który sugerował, iż Anktiva, preparat zatwierdzony do leczenia raka pęcherza, może leczyć wszystkie nowotwory i działać jak szczepionka przeciw rakowi, nie znajduje potwierdzenia w danych klinicznych.
To trzecia reakcja FDA na poczynania władz ImmunityBio, po tym, gdy na podobne problemy promocyjne urząd wskazywał w korespondencji wysłanej do spółki we wrześniu 2025 i styczniu 2026.
ImmunityBio ma 15 dni roboczych na formalną odpowiedź na pismo ostrzegawcze nadzorcy, w którym powinna przedstawić plan usunięcia naruszeń oraz przekazać odbiorcom wprowadzających w błąd materiałów promocyjnych stosowne komunikaty korygujące.
- EMA wyraziła obawy, że leki onkologiczne Baxter International zawierające ifosfamid pozostaną w niedoborze w całej Unii Europejskiej aż do przyszłego roku. Są one sprzedawane na terenie Wspólnoty m. in. pod nazwami handlowymi Holoxan, Tronoxal i Mitoxana.
Regulator poinformował, że zidentyfikowany problem wynika z usterki technicznej oraz konieczności modernizacji zakładu po kontroli regulacyjnej, co spowodowało tymczasowe wstrzymanie produkcji. Po inspekcji wytwarzanie ifosamidu zostało wznowione, lecz fabryka ma osiągnąć pełną zdolność produkcyjną dopiero w pierwszym kwartale 2027, co ma wpływ na wszystkie państwa UE i EOG.
Mając to na uwadze EMA opublikowała zalecenia dla pracowników ochrony zdrowia, którzy powinni ocenić, czy mają wystarczające zapasy leku, aby ukończyć pełny cykl terapii zanim rozpoczną leczenie nowych pacjentów za pomocą ifosfamidu. Gdyby dostrzegli niedobór, to powinni rozważyć przejście pacjentów na alternatywne terapie zgodnie z właściwymi wytycznymi klinicznymi.
- Aardvark Therapeutics dobrowolnie zawiesił badania II fazy klinicznej POWER i STRENGTH dla kandydata na lek przeciwko otyłości ARD-201 z powodu niepokojących sygnałów dotyczących bezpieczeństwa. To drugi cios w pipeline, gdyż niedawno spółka odłożyła na półkę badanie ARD-101 w zespole Prader-Williego.
ARD-201 była testowana wśród pacjentów, którym udało się stracić 15% masy ciała, pod kątem utrzymania trwałości regresu wagi (nazywa się to czasami efektem jojo następującym po zaprzestaniu preparatów GLP-1). Aardvark podał, że u dwóch pacjentów uczestniczących w próbie II fazy klinicznej wystąpiły anomalie echokardiograficzne, które mogą wskazywać na obniżoną wydajność serca. Firma kontynuuje ocenę tych ustaleń w dziedzinie bezpieczeństwa, współpracując z FDA, a aktualizacją z procesu podzieli się z publicznością w II kw. 2026.
- Karyopharm Therapeutics opublikował wyniki III fazy klinicznej dla terapii ukierunkowanej na mielofibrozę (nowotwór szpiku). Spółka ogłosiła, że badanie SENTRY, oceniające selineksor w skojarzeniu z ruksolitynibem w pacjentów z tym nowotworem dowiodło istotnego zmniejszenia objętości śledziony.
Próba wykazała, że 50% pacjentów otrzymujących terapię z selineksorem osiągnęło redukcję objętości śledziony o co najmniej 35% po 24 tygodniach w porównaniu z 28% pacjentów leczonych wyłącznie ruksolitynibem. Odsetek pacjentów przerywających leczenie z powodu działań niepożądanych wynosił 15% w kohorcie terapii skojarzonej (selineksor/ruksolitynib) wobec 9% w grupie otrzymującej wyłącznie ruksolitynib.
Środa (25.3.2026)
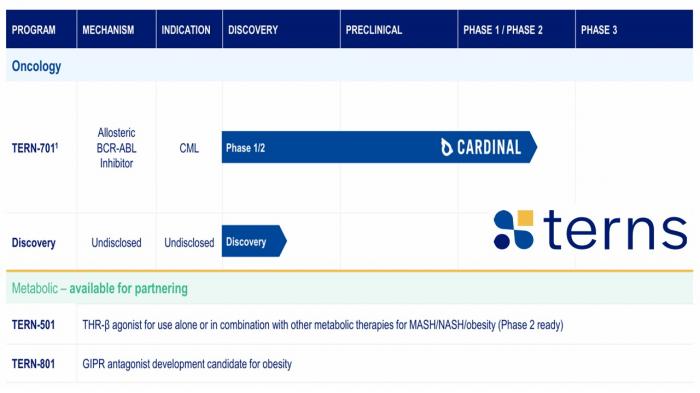
- Merck wyda do 6,7 mld dol. na Terns Pharma. Dzięki przejęciu Merck zdobędzie TERN-701, głównego kandydata na lek celu transakcji, który jest testowany pod kątem przewlekłej białaczki szpikowej.
"Przejęcie Terns rozwija naszą rosnącą pozycję w hematologii dzięki TERN-701, potencjalnemu kandydatowi najwyższej klasy do leczenia niektórych pacjentów z przewlekłą białaczką szpikową. Ta transakcja dodatkowo dywersyfikuje i wzmacnia naszą pozycję w onkologii, gdy nadal poszukujemy możliwości rozszerzenia naszego portfolio na inne obszary terapeutyczne” – powiedział Robert M. Davis, dyrektor generalny Merck.
Operacja, która dobrze wpisuje się w poszukiwanie obiecujących cząsteczek w obszarze onkologii ze względu na zbliżający się w 2028 r. klif patentowy Keytrudy, zapewniła 31% premię w stosunku do 60-dniowej średniej ważonej wolumenem ceny akcji Terns (Merck kupi papiery po 53 dol. za sztukę).
Analitycy BMO Capital Markets uznali, że przejęcie Terns, to jedna z najlepszych transakcji, jakich Merck dokonał w związku z oczekiwanym ubytkiem sprzedaży Keytrudy po zmierzeniu się z konkurencją ze strony producentów leków biopodobnych. Ich zdaniem TERN-701 stanie się blockbusterem, którego sprzedaż w szczycie będzie mierzona w miliardach dolarów, co pozwoli w znaczący sposób załatać dziurę sprzedażową z powodu wygaśnięcia wyłączności patentowej Keytrudy.
W grudniu 2025 Terns Pharma zaprezentowała odczyt wyników I fazy klinicznej, które wykazały ogólny wskaźnik odpowiedzi molekularnej sięgający 75% w 24. tygodniu podawania kandydata na lek z profilem tolerancji wspierającym codzienne dawkowanie.
"Bezprecedensowy pozostaje jedynym odpowiednim przymiotnikiem opisującym profil kliniczny tego związku” - ocenili 9 grudnia 2025 w notce dla klientów jakość preparatu analitycy banku inwestycyjnego William Blair.
Według nich TERN-701 jest na dobrej drodze, by zakwestionować dominację Scembliksu i zakłócić paradygmat leczenia białaczki szpikowej. Zapewne dopuszczenie TERN-701 stanowiłoby poważne zagrożenie dla Novartisu, którego Scemblix w październiku 2021 uzyskał dopuszczenie, dzięki czemu wygenerował w zeszłym roku 1,285 mld dol. sprzedaży.
Co więcej, eksperci William Blair sądzą, że kwota transakcji nie oddaje w pełni potencjału sprzedażowego TERN-701. W związku z tym spodziewają się, że w wyścigu o Terns może pojawić się konkurent. Podobnej opinii byli finansiści RBC Capital Markets, którzy wskazali, że w gronie rywali Mercka mogą pojawić się inni przedstawiciele Big Pharmy. Pod względem sensu strategicznego transakcja przejęcia Terns najbardziej pasowałaby AbbVie lub Bristol Myers Squibb.
Źródło: Terns Pharma
- FDA przyznała zgodę na wprowadzenie do obrotu relacorilantu, leku do terapii agresywnej postaci raka jajnika (niereagującego już na standardową terapię), który Corcept Therapeutics będzie sprzedawał pod nazwą handlową Lifyorli. Lek ma być stosowany wraz z nab-paklitakselem u dorosłych kobiet z nabłonkowym rakiem jajnika, jajowodu lub otrzewnej pierwotnej opornym na platynę. Lifyorli jest pierwszym selektywnym antagonistą receptora glukokortykoidowego dopuszczonym przez FDA.
Werdykt agencji bazował na wynikach badania III fazy klinicznej ROSELLA. Wykazało ono, że pacjentki leczone Lifyorli w skojarzeniu z nab-paklitakselem miały o 35% mniejsze ryzyko zgonu w porównaniu z osobami otrzymującymi wyłącznie nab-paklitaksel, a mediana całkowitego przeżycia wydłużyła się o 4,1 miesiąca.
Decyzja była zaskoczeniem z powodu terminu, gdyż zapadła około 4 miesiące wcześniej niż zakładali analitycy sektora ochrony zdrowia. Dzięki niej portfel skomercjalizowanych leków Corceptu powiększy się – do tej pory jednym takim preparatem był Korlym, lek do terapii hiperkortyzolizmu. Na pierwszej sesji po ogłoszeniu sukcesu regulacyjnego akcje Corcept Therpeutics zyskały 19,7%.
Czwartek (26.3.2026)
- FDA zatwierdziła icodec (Awiqli) Novo Nordisk, zastrzyk insulinowy, który poprawia poziom cukru we krwi u dorosłych z cukrzycą typu 2. To pierwsza dopuszczona do dawkowania raz w tygodniu insulina bazowa, dzięki czemu ograniczy się znacząco częstość podawania (z siedmiu razy w tygodniu do jednego razu w tym okresie).
Regulator oparł decyzję na wynikach badania fazy klinicznej IIIa ONWARDS, w którym wzięło udział 2 680 osób. Próba wykazała wystarczającą skuteczność i bezpieczeństwo icodeku w porównaniu z innymi insulinami bazowymi.
- Wave Life Sciences zaliczył nieudaną sesję, gdyż akcje spółki zniżkowały o 49,6% po tym, gdy do inwestorów dotarły wieści o rezultatach z badania INLIGHT dla WVE-007 w I fazie klinicznej, kandydata na lek w terapii otyłości. WVE-007 to terapia z użyciem małych interferujących RNA (siRNA), zaprojektowana do wyciszania genu INHBE związanego z magazynowaniem tłuszczu. Wave zaprojektował mechanizm działania kandydata na lek tak, aby chronił mięśnie, co jest częstym problemem w lekach GLP-1, i wymagał rzadszego dawkowania niż obecnie dostępne terapie.
Dane z sześciomiesięcznej oceny pojedynczej dawki 240 mg wykazały wprawdzie uwidoczniły 14,3% redukcję tkanki tłuszczu trzewnego, lecz wyższa dawka (400 mg) przyniosła spadek 0,9%, a analitycy oczekiwali 2-3%.
W grudniu ubiegłego roku spółka upubliczniła dane po 3 miesiącach stosowania dawki 240 mg, które wykazały 9,2% spadek tłuszczu trzewnego, a analitycy Truist Securities określili je jako imponujące, co doprowadziło do 80% wzrostu ceny giełdowej. Tym razem, głęboka przecena pokazuje obraz rozczarowania inwestorów, lecz specjaliści branżowi dostrzegli iskierkę nadziei w najświeższych danych.
Cheng Li, analityk Oppenheimera, napisał, że warto wykorzystać każdą słabość ceny giełdowej, ponieważ opublikowane rezultaty potwierdzają czysty profil bezpieczeństwa oraz dawkowanie WVE-007 raz lub dwa razy w roku.
Spółka przy prezentacji wyników posiłkowała się wskaźnikiem masy tłuszczu trzewnego do mięśni trzewnych dla dawki 240 mg po 6 miesiącach aplikowania. Według obliczeń Wave relacja obu parametrów dla jego kandydata na lek spadła o 16,5% w stosunku do wartości wyjściowej, czyli lepiej niż w przypadku semaglutydu Novo Nordisk (12,2%).
Mizuho Securities oceniło najnowszy odczyt dla WVE-007 jako dobry, a jego uwagę zwróciło zmniejszenie tłuszczu trzewnego i obwodu talii przy jednoczesnym stabilizowaniu masy czystej, co było szczególnie zachęcające i wciąż pokazuje WVE-007 jako zróżnicowany mechanizm terapii otyłości. Japońska firma inwestycyjna uważa, że sprzedaż globalna WVE-003 w szczycie wyniesie ok. 7 mld dol.
- Kodiak Sciences podał, że Zenkuda (tarcocimab tedromer) testowana w badaniu III fazy klinicznej GLOW2 osiągnęła główny cel u pacjentów z uszkodzeniem wzroku związanym z cukrzycą.
Próba wykazała, że 62,5% pacjentów leczonych Zenkudą osiągnęło znaczną, co najmniej dwustopniową poprawę w skali nasilenia retinopatii cukrzycowej w porównaniu z jedynie 3,3% pacjentów otrzymujących placebo. Spełniony został też kluczowy drugorzędowy punkt końcowy badania – doszło do 85% redukcji ryzyka wystąpienia powikłań zagrażających widzeniu u pacjentów leczonych Zenkudą. Kandydat na lek był dobrze tolerowany i miał korzystny profil bezpieczeństwa (brak przypadków zapalenia wewnątrzgałkowego).
Inwestorzy zareagowali optymistycznie na publikację wyników, ponieważ w czwartek poszły w górę o niemal 75%.
Piątek (27.3.2026)
- Sanofi dostała od EMA rekomendację zatwierdzenie podskórnej wersji Sarclisy (isatuksimab), która służy terapii szpiczaka mnogiego. Gdyby zapadła decyzja zgodna z zaleceniem, to Sarclisa stałaby się pierwszym w UE lekiem przeciwnowotworowym podawanym przez urządzenie noszone na ciele.
System Sanofi opracowany przy współudziale Enable Injections ma szansę zapewnić wygodę leczenia, ponieważ jego podanie zabierałoby kilka minut w porównaniu z wielogodzinnymi dożylnymi wlewami Sarclisy jak obecnie. Branża spodziewa się, że ostateczna decyzja unijnego regulatora zapadnie w ciągu najbliższych kilku miesięcy.
- Medtronic dostał zgodę FDA na dopuszczenie do obrotu systemu chirurgicznego Stealth AXiS, który może mieć zastosowanie w operacjach czaszki, ucha, nosa i gardła, co rozszerza wskazanie dla platformy. Stealth AXiS jest platformą łączącą planowanie, nawigację i robotykę w jeden spójny system. W lutym br. FDA dała zgodę na jej stosowanie w procedurach medycznych dotyczących kręgosłupa.
- FDA zatwierdziła terapię genową Rocket Pharmaceuticals za pomocą Kresladi (marnetegragene autotemcel), która służy leczeniu ciężkiego niedoboru adhezji leukocytów typu I (LAD-I), spowodowanego mutacjami w genie ITGB2.
Kresladi jest terapią jednej dawki, przeznaczonej dla dzieci, które nie mają zgodnego dawcy rodzeństwa do przeszczepu komórek macierzystych. Po decyzji amerykańskiego regulatora można spodziewać się, że w najbliższym czasie wniosek o zatwierdzenie Kresladi trafi również do unijnej EMA.
- AstraZeneca podała, że tozorakimab osiągnął główny cel w dwóch badaniach (OBERON i TITANIA) III fazy klinicznej, wykazując istotne zmniejszenie liczby zaostrzeń przewlekłej obturacyjnej choroby płuc (POChP). Kandydat na lek był generalnie dobrze tolerowany i miał korzystny profil bezpieczeństwa. Jego stosowanie doprowadziło do zmniejszenia rocznej częstości umiarkowanych do ciężkich zaostrzeń POChP w porównaniu z placebo, w pierwotnej populacji byłych palaczy oraz w całej populacji, obejmującej byłych i obecnych palaczy.
Cząsteczką jest potencjalnie pierwszym w swojej klasie przeciwciałem monoklonalnym skierowanym na interleukinę-33 (IL-33), które unikalnie hamuje sygnalizację zredukowanych i utlenionych form IL-33, oferując potencjał zarówno do redukcji stanu zapalnego, jak i zakłócenia cyklu dysfunkcji śluzu przyczyniającego się do pogorszenia POChP.
"Dzisiejsze wyniki tozorakimabu dostarczają pierwszych dwóch potwierdzających badań fazy III leku biologicznego IL-33, który jest znaczącym postępem naukowym w dziedzinie POChP, trzeciej na świecie przyczyny zgonów. Tozorakimab działa zasadniczo inaczej niż inne leki biologiczne, hamując sygnalizację zredukowanych i utlenionych form IL-33, aby zarówno zmniejszyć stan zapalny, jak i zakłócić cykl zaburzeń śluzu, które są kluczowymi czynnikami chorób POChP" - oceniła Sharon Barr, wiceprezes wykonawcza ds. badań i rozwoju biofarmaceutyków AstraZeneca.
- Novartis nabędzie za do 2 mld dol. kalifornijską spółkę biotechnologiczną Excellergy. Cel przejęcia ma swoim portfelu badawczym Exl-111, nowej generacji przeciwciało anty-IgE, kandydata na lek w terapii chorób alergicznych, który znajduje się w badaniu I fazy klinicznej. Strony transakcji zamierzają sfinalizować ją w II połowie tego roku.
Obserwuj nas na  Google News
Google News
Chcesz być na bieżąco z wieściami z naszego portalu? Obserwuj nas na Google News!

Reklama
Komentarze opinie
Podziel się swoją opinią
Twoje zdanie jest ważne jednak nie może ranić innych osób lub grup.





Reklama
Reklama




Reklama
Reklama
Reklama
Najnowsze wiadomości
- 18/07 Telemedycyna dla seniorów: co oferuje NFZ?
- 18/07 Wyrabiamy kartę EKUZ bez wychodzenia z domu. Zajmuje to tylko kilka minut
- 18/07 AOTMiT o otyłości. Kompleksowa opieka bariatryczna bliżej koszyka świadczeń
- 17/07 Symfonia życia w Kajetanach. Co muzyka robi z mózgiem osoby z implantem ślimakowym?
- 17/07 Kriomedpol zmienia zarząd. Nowe otwarcie dla producenta urządzeń do krioterapii i kriochirurgii
- 17/07 Prawie 16 tys. kandydatów na WUM. Uczelnia: „Jesteśmy liderem pod względem bezwzględnej liczby kandydatów”
- 17/07 NIK: lekarz pracował 144 godziny bez przerwy. Kontrola SOR-ów ujawniła przeciążenie personelu i systemowe braki
- 17/07 „Setki milionów złotych miesięcznie zagrożone”. PAŻP walczy o odblokowanie środków, jest formalny sprzeciw do sądu w Brukseli
- 17/07 Kadry. Ppłk Agata Będzichowska została Komendantem CSK MON WIM-PIB
- 17/07 Jest podpis prezydenta. Kiedy wystartuje Bon Senioralny?
- 17/07 Dietetycy dostaną własną ustawę. Wioleta Tomczak: Zachęcam do konsultacji publicznych
- 17/07 Kotula po wecie ustawy o statusie osoby najbliższej: „To nie blokada, a opóźnienie zmiany prawa”
- 17/07 Sześć nowych projektów dla ochrony zdrowia. „Obowiązek przepracowania przynajmniej pół etatu w jednym szpitalu”
- 17/07 Prezydent zawetował ustawę o statusie osoby najbliższej. „Nie podpiszę alternatywy dla małżeństwa”. Podpisane dwie inne ustawy
- 17/07 Pokonali raka, ale stracili głos. Onkologopeda: rehabilitacja powinna zacząć się przed leczeniem
- 17/07 Mniej niż 20 proc. dzieci jest wystarczająco aktywnych. Dr Marta Łabęcka: kondycję fizyczną przedszkolaków trzeba diagnozować wcześniej
- 16/07 „Bez dobrej diety nie będzie progresu”. Dietetyk: wyniki sportowe zaczynają się w kuchni
- 16/07 Taka ankieta przed rezonansem to za mało. Rzecznik Praw Pacjenta upomina
- 16/07 Ochłodzenie skóry pomogło pacjentkom zachować włosy podczas chemioterapii
- 16/07 E-skierowanie: jak działa i jak korzystać?
Komentarze