
Reklama
Brytyjski koncern Big Pharmy szarpnął się na największy wydatek na przejęcie Nuvalent Bio od ponad dekady, na co przeznaczy 10,6 mld dol. Dzięki temu wzmocni swoje portfolio onkologiczne, ponieważ doda do niego znajdujące się tuż przed zatwierdzeniem zidesamtinib i neladalkib, kandydatów na leki w niedrobnokomórkowym raku płuc. Sypnęło odczytami w terapiach nadwagi/otyłości - wyniki AstryZeneki dla elecoglipronu (II faza) i Zealand Pharmy dla survodutide (III faza) przejęto z rozczarowaniem, a Structure Therapeutics dla aleniglipronu (II faza) uznano za obiecujące. Zasocitinib Takedy wykazał wyższość w terapii łuszczycy w porównaniu z Sotyktu Bristol Myers Squibb. Novartis pochwalił się odczytem I/II fazy klinicznej dla del-braksu, preparatu testowanego u cierpiących na dystrofię mięśniową w odmianie twarzowo-łopatkowo-ramieniowej.
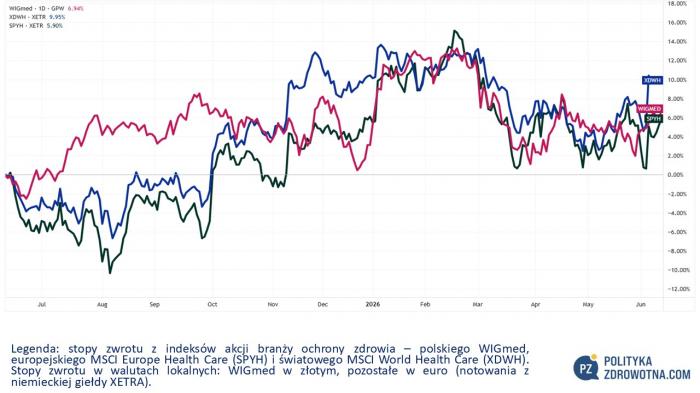
WIGmed poszedł w górę o 1,5% w ostatnim tygodniu. Europejskie akcje spółek sektora ochrony zdrowia uwzględniane w indeksie MSCI Europe Health Care (SPYH) poszły dół o 0,4%. Globalny indeks MSCI World Health Care (XDWH) zyskał 0,8%.
Źródło: TradingView
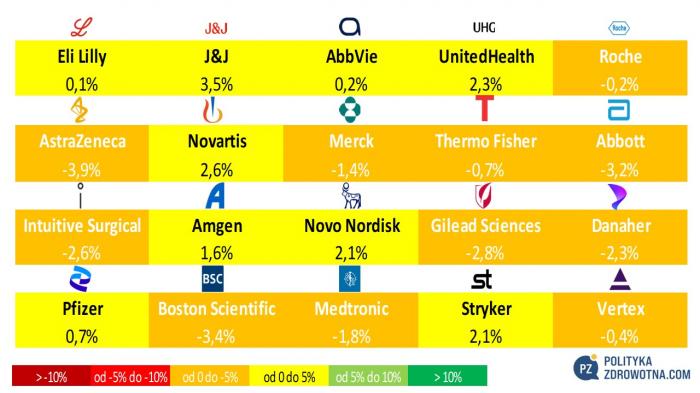
W wiodącej dwudziestce największych firm z MSCI World Health Care wyróżniły się pozytywnie przede wszystkim akcje Johnson & Johnson (+3,5%), UnitedHealth Group (+2,3%) i Novo Nordisk oraz Stryker ez-aequo (+2,1%). Najsłabsze okazały się papiery AstraZeneca (-3,9%), Boston Scientific (-3,4%) i Abbott (-3,2%).
Reklama
Źródło: opracowanie własne na podstawie stooq.pl
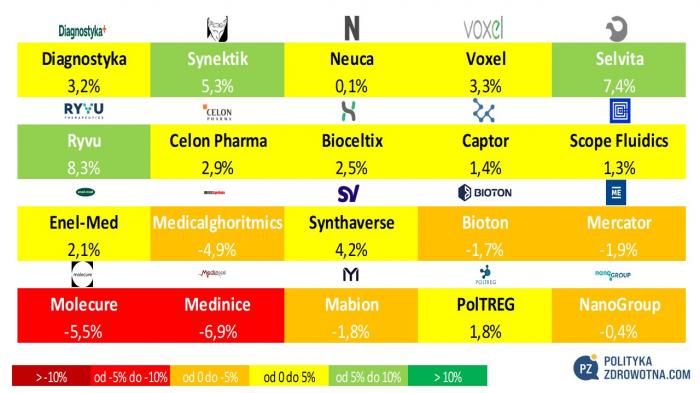
W Polsce w gronie najlepiej radzących sobie na parkiecie znalazły się akcje Ryvu Therapeutics (+8,3%), Selvity (+7,4%) i Synektik (+5,3%). Największe straty odnotowali posiadacze walorów Medinice (-6,9%), Molecure (-5,5%) i Medicalghoritmics (-4,9%).
Źródło: opracowanie własne na podstawie stooq.pl
Działo się w Polsce
Poniedziałek (8.6.2026)
- Celon Pharma zawarła umowę z Pharmalink Drug Store w celu komercjalizacji preparatów semaglutydu w wielodawkowych penach do wstrzyknięć podskórnych. Porozumienie dotyczy leku Reduzek w Arabii Saudyjskiej, Zjednoczonych Emiratach Arabskich, Kuwejcie, Bahrajnie, Katarze i Omanie. Minimalna wartość umowy w okresie pierwszych trzech pełnych lat następujących po pierwszej sprzedaży komercyjnej wynosi 29 mln dol., na co składają się płatności z tytułu osiągnięcia kamieni milowych oraz przychody z dostaw. Umowa obowiązuje przez okres 5 lat od pierwszej sprzedaży produktu w jednym ze wskazanych państw.
Reklama
"Celon Pharma pozostanie odpowiedzialny za dostawy produktów, dokumentację rejestracyjną oraz wsparcie produkcyjne. Pharmalink będzie odpowiedzialny za lokalną rejestrację, import, dystrybucję, marketing, sprzedaż, nadzór nad bezpieczeństwem farmakoterapii i działania związane z komercjalizacją na wskazanym terytorium" - wyjaśniła spółka.
Współpraca obejmuje dwa pakiety dokumentacji rejestracyjnej produktów: Reduzek 0,25 mg, 0,5 mg, 1,0 mg i 2,0 mg do leczenia cukrzycy typu 2 i kontroli glikemii oraz Reduzek 0,25 mg, 0,5 mg, 1,0 mg, 1,7 mg i 2,4 mg do przewlekłego kontrolowania masy ciała/otyłości.
Reklama
Wtorek (9.6.2026)
- Good For You Medical, podmiot przejmowany przez MedTech Solutions, sprzedał system obrazowania śródoperacyjnego Phoenix 3000 w 10 Wojskowym Szpitalu Klinicznym wraz z Polikliniką SPZOZ w Bydgoszczy. Pozostałe szczegóły umowy dostawy są nieznane.
"Systematycznie przechodzimy z fazy budowy portfolio do fazy przyspieszonej komercjalizacji technologii medycznych. Każde kolejne wdrożenie w kluczowych ośrodkach klinicznych zwiększa przewidywalność przychodów, wzmacnia pipeline sprzedażowy oraz potwierdza zdolność Grupy do skalowania działalności w segmentach o wysokiej barierze wejścia. Phoenix 3000 jest elementem szerszej strategii budowy portfela technologii o powtarzalnym potencjale wdrożeniowym" - zaznaczył Jarosław Kaim, prezes zarządu MedTech Solutions.
Reklama
Środa (10.6.2026)
- Urteste włączyło pierwszego uczestnika do badania klinicznego testu Panuri w diagnostyce raka trzustki. Spółka spodziewa się wyników analizy pośredniej (interim analysis) w IV kwartale 2026.
"Włączenie pierwszego pacjenta do badania klinicznego z wykorzystaniem testu Panuri to dla nas ważny krok w kierunku komercjalizacji. Zakładamy sprawny przebieg rekrutacji i oczekujemy pierwszych wyników analizy pośredniej jeszcze pod koniec roku. Uzyskanie pozytywnych wyników istotnie zwiększy prawdopodobieństwo zawarcia transakcji partneringowej. Zainteresowanie testem Panuri jest bardzo duże. Panuri jest rozwijany jako nieinwazyjne, skuteczne i przystępne cenowo narzędzie wspierające wczesną diagnostykę raka trzustki - jednego z najpóźniej rozpoznawanych i najgorzej rokujących nowotworów" - powiedział Grzegorz Stefański, prezes zarządu Urteste.
Reklama
Głównym celem badania jest ocena skuteczności testu Panuri w wykrywaniu raka trzustki, przy czym pierwszorzędowe punkty końcowe obejmują czułość i swoistość testu.
- Medicalgorithmics zwołał na 15 lipca br. kolejne walne zgromadzenie w sprawie emisji akcji serii P, które miałby zostać objęte przez Biofund Capital Management po cenie emisyjnej 33 zł za papier (w piątek na zamknięciu sesji kosztowały one 26,4 zł).
- Biofund Capital Management, główny inwestor projektu multiQure, podpisał umowę inwestycyjną z Pure Biologics, w ramach której zobowiązał się do wprowadzenia aportem do spółki platformy RNAi, w zamian za co dostanie nowe akcje Pure, które będą stanowiły docelowo 49,99% udziału w spółce.
Reklama
Ponadto Pure Biologics w ramach umowy zobowiązał się do wyemitowania do 50 mln nowych akcji, aby pozyskać środki na rozwój projektu multiQure. Firma podpisała też umowę konwersji z ACRX, swoim kluczowym akcjonariuszem, który zobowiązał się do konwersji pożyczki udzielonej Pure Biologics w zamian za akcje.
Decyzję w sprawie emisji akcji podjęło walne zgromadzenie akcjonariuszy 10 czerwca br. Pure zmieni nazwę na multiQure, co zwieńczy proces przekształcenia.
"Umowa Biofund z Pure Biologics to dla nas przełomowy moment, który otwiera przed projektem multiQure nową, ambitną przyszłość. Już dziś akcjonariusze spółki podejmą kluczową decyzję, która będzie istotnym driverem dalszego rozwoju projektu. Wierzymy, że wniesienie platformy multiQure do już notowanego podmiotu pozwoli nie tylko w pełni wykorzystać jej potencjał, ale przede wszystkim zapewni dotychczasowym oraz nowym akcjonariuszom Pure Biologics silną ekspozycję na światowy rynek terapii genowych przeciwko chorobie Huntingtona, którego potencjał szacowany jest na dziesiątki miliardów dolarów. To szansa na budowę wartości dla inwestorów w obszarze, w którym zapotrzebowanie na skuteczne terapie jest ogromne" - powiedział Tomasz Nocuń, szef projektu multiQure.
Reklama
- Bioceltix dostał pozwolenie na budowę wytwórni biologicznych leków weterynaryjnych we Wrocławiu, co obejmie m. in. przebudowę fragmentu budynku hali magazynowej z zapleczem socjalno-biurowym na potrzeby laboratoriów z zapleczami oraz zmianę sposobu użytkowania części pomieszczeń na potrzeby wytwórni biologicznych leków weterynaryjnych.
Czwartek (11.6.2026)
- Synektik oczekuje dalszej poprawy wyników finansowych w II półroczu trwającego roku obrotowego. Spółka, będąca dystrybutorem regionalnym systemów robotycznych, planuje zwiększyć udział przychodów powtarzalnych do 60% w ciągu najbliższych 2-3 lat (obecnie jest to nieco poniżej 50%).
Reklama
Działo się na świecie
Poniedziałek (8.6.2026)
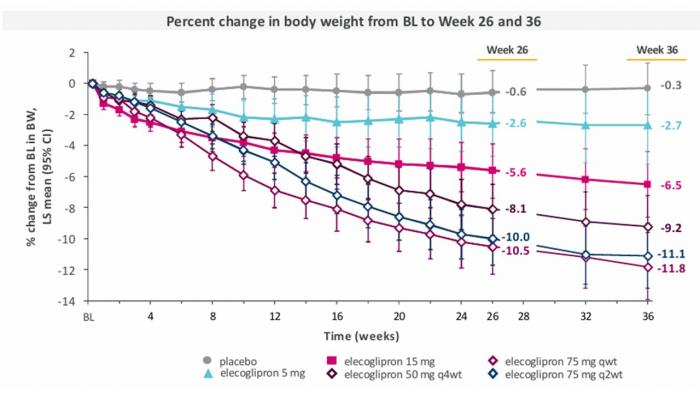
- AstraZeneca poinformowała, że pacjenci przyjmujący raz dziennie tabletkę 75 mg elecoglipronu (ECC5004) stracili 10,5% masy ciała po 26. tygodniach badania II fazy klinicznej VISTA (w grupie placebo było 0,6%). Spółka ujawniła też podczas zjazdu American Diabetes Association (ADA), że po 36. tygodniach utrata masy ciała sięgnęła 11,8% (placebo 0,3%).
Elecoglipron wykazał również klinicznie istotną poprawę w kilku analizach eksploracyjnych czynników ryzyka kardiometabolicznego, w tym w zakresie obniżenia ciśnienia krwi i poziomu białka C-reaktywnego, będącego markerem stanów zapalnych ogólnoustrojowych.
Reklama
W związku z tym brytyjsko-szwedzki koncern BigPharmy planuje rozpocząć III fazę kliniczną. Program rozwoju III fazy obejmuje badanie EMBOLD, które oceni cząsteczkę pod kątem leczenia osób z otyłością lub nadwagą, z cukrzycą typu 2, jak i bez niej, i badanie ELUMINATE, analizujące ją jako monoterapię oraz w połączeniu z dapagliflozyną. Dodatkowe badania będą koncentrować się na długoterminowych wynikach sercowo-naczyniowych i nerek.
"Naszym celem jest posiadanie portfolio, które potrafi zwalczać wiele chorób sercowo-naczyniowych, nerkowych i metabolicznych, będących głównymi przyczynami zgonów na całym świecie. Rozwój elecoglipronu to ważny krok w realizacji zróżnicowanego portfolio zarządzania masą ciała, oferującego monoterapie i kombinacje, zaprojektowane do rozwiązania biologicznej złożoności otyłości i chorób współistniejących, które można dostosować do indywidualnych potrzeb, umożliwiając ludziom zdrowsze życie. Te wyniki dają nam pewność, żeby rozpocząć nasz rozległy program fazy III” - wyznała Sahron Barr, wiceprezes wykonawcza ds. rozwoju biofarmaceutyków AstraZeneki.
Reklama
Firma weszła w posiadanie elecoglipronu w ramach licencji od chińskiej Eccogene, uzyskanej pod koniec 2023 r. o potencjalnej wartości 2 mld dol. Dzięki osiągnięciu klinicznemu Astra ma szansę wejść do rywalizacji z Novo Nordisk i Eli Lilly w segmencie doustnych terapii leczenia nadwagi/otyłości, choć ich preparaty cechuje nieco wyższa skuteczność (Novo 14%, Lilly 12% redukcji masy ciała).
Analitycy banku inwestycyjnego BMO Capital Markets określili rezultaty Astry jako stosunkowo rozczarowujące, zwłaszcza w kontekście odczytu podanego przez Structure Therapeutics podczas spotkania ADA - link: https://structuretx.com/wp-content/uploads/2026/06/ADA-2026-poster-ACCESS-II-v3-for-upload.pdf.
Structure ujawnił, że testowany przez niego aleniglipron w środkowej fazie klinicznej cechował 15,3% utrata masy ciała skorygowanej przez placebo w 36. tygodniu, gdy dawka 120 mg została podwyższona do 240 mg (w przypadku 120 mg redukcja wyniosła 11,3%).
Według ekspertów BMO AstraZeneca może mieć niewielką przewagę pod względem wskaźników bezpieczeństwa w porównaniu ze Structure, ale różnica ta może nie być wystarczająca.
"Chociaż wskaźniki nudności i działań niepożądanych prowadzących do przerwania leczenia są nieco niższe, nie uważamy tej różnicy za szczególnie istotną" - ocenili specjaliści kanadyjskiej instytucji finansowej.
Źródło: AstraZeneca
- Johnson & Johnson przejmie Firefly Bio za 1 mld dol. płatne gotówką. Dzięki transakcji globalny numer 2 w segmencie healthcare pod względem kapitalizacji giełdowej (580 mld dol., co stanowi 57% wartości rynkowej liderującego Eli Lilly) uzyska dostęp do innowacyjnej platformy nabywanego podmiotu - Firelink, która koncentruje się na opracowywaniu nowej klasy terapii onkologicznych zwanych Degrader Antibody Conjugates (DAC).
Technologia łączy precyzję koniugatów przeciwciało-lek (ADC) z katalityczną mocą degraderów białkowych. Firelink jest ukierunkowana szczególnie na nowotwory z mutacją w genie KRAS.
- Incyte uzgodniło przejęcie Vega Therapeutics, będącej własnością Star Therapeutics, w transakcji wartej do 2 mld dol. Spółka zapłaci za kupowane przedsiębiorstwo 1,25 mld dol. z góry, a do tego dochodzi 750 mln dol., których wypłata zależy od spełnienia kamieni milowych rozwojowych i komercyjnych. Dzięki operacji Incyte wzmacnia swoje portfolio w dziedzinie hematologii.
Wiodącym programem badawczym Vegi jest terapia VGA039, skierowana przeciwko chorobie von Willebranda, najczęstszym wrodzonym zaburzeniu krzepnięcia krwi. Znaduje się ona w III fazie klinicznej (badanie VIWID).
- FDA zatwierdziła BeneFIX Pfizera, dożylną sztuczną wersję czynnika krzepnięcia IX, która jest przeznaczona do zapobiegania lub ograniczania częstości epizodów krwawienia u dzieci oraz u niektórych pacjentów z hemofilią B.
Amerykański nadzorca rozszerzył etykietę preparatu o wskazanie pediatryczne, gdyż BeneFIX został po raz pierwszy dopuszczony do obrotu w 1997 r., stając się pierwszą rekombinowaną terapią czynnikiem IX dostępną w USA w leczeniu hemofilii B.
- Roche zawarł wyłączne porozumienie licencyjno-badawcze Nurix Therapeutics o wartości do 2,3 mld dol. Umowa koncentruje się na bexobrutidegu (NX-5948), degraderze kinazy tyrozynowej Brutona (BTK) Nuriksa. Partnerzy planują wprowadzić cząsteczkę do badania III fazy klinicznej w leczeniu przewlekłej białaczki limfocytowej latem tego roku, a ponadto przeanalizować jego potencjał w obszarze immunologii i neurologii.
Szwajcarski koncern z Big Pharmy zapłaci z góry gotówką 700 mln dol. Reszta wypłaty jest uzależniona od osiągnięcia potencjalnych kamieni milowych w zakresie rozwoju, regulacji i sprzedaży. Spółki porozumiały się co do podziału kosztów rozwoju - Roche pokryje 60% z nich, a pozostałą część Nurix.
- Zealand Pharma nie dał akcjonariuszom powodów do radości po publikacji wyników badania III fazy klinicznej survodutide, cząsteczki współrozwijanej z niemieckim Boehringer Ingelheim, ukierunkowanej na leczenie otyłości. Na wieść o rezultatach kurs akcji duńskiej spółki zniżkował 19,1%. Powodem do negatywnej reakcji inwestorów były gorsze skutki uboczne i wyższy wskaźnik rezygnacji z przyjmowania terapii przez pacjentów niż u konkurencyjnych leków.
Spółka podała, że blisko co piąty pacjent zaprzestał terapii, co okazało się dużo wyższym wskaźnikiem rezygnacji niż w kohorcie placebo (2,9%). Najczęściej zgłaszanymi działaniami niepożądanymi były nudności, wymioty i biegunka, typowe dla tej klasy leków, ale występowały one częściej niż w konkurencyjnych cząsteczkach.
Survodutide mimo problemów z tolerancją spełnił jednak główne cele badania. Znacząco zmniejszył ilość tłuszczu trzewnego i wątrobowego przy jednoczesnym ograniczeniu utraty beztłuszczowej masy mięśniowej. Po 76. tygodniach spadek tłuszczu trzewnego sięgnął 34%, zaś wątrobowego 63,1%.
Wtorek (9.6.2026)
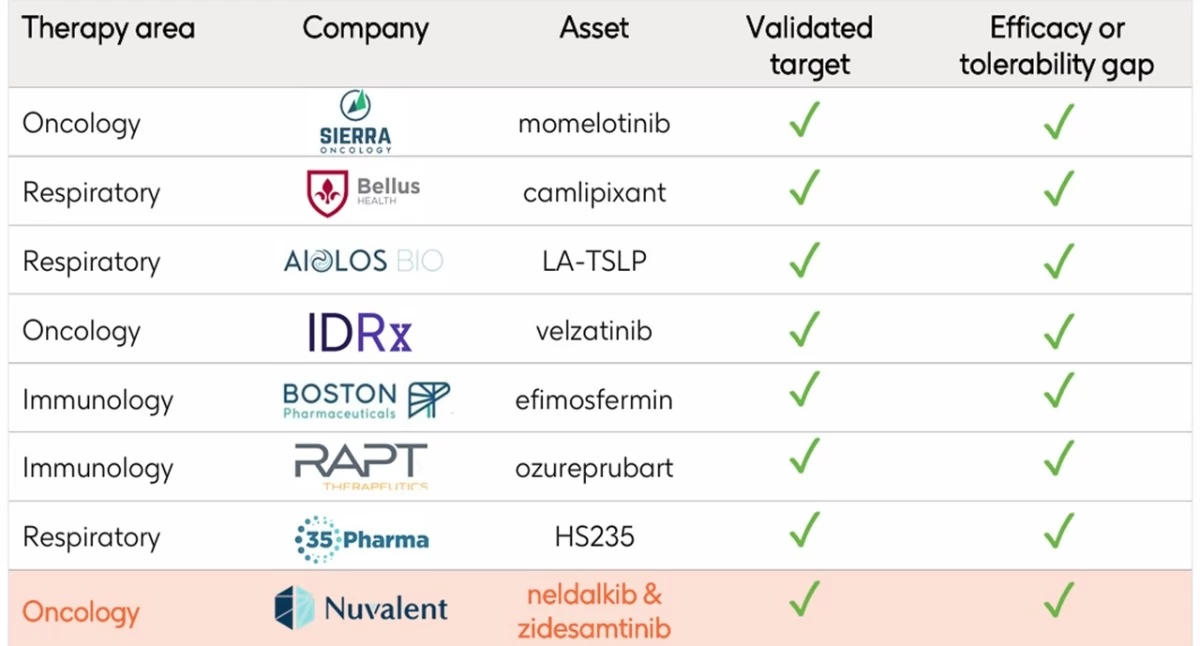
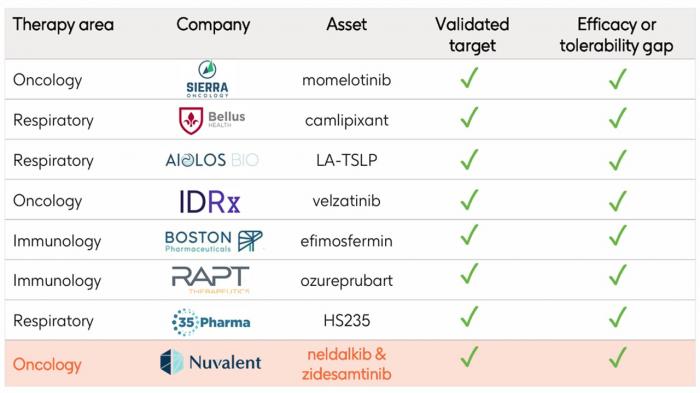
- GSK przejmie Nuvalent Bio za 10,6 mld dol., co jest drugą największą wartościowo transakcją w tym roku (po kwietniowym dealu Sun Pharmy, która wyłożyła 11,75 mld dol. w kwietniu na Organon). Dla GSK to trzeci duży tegoroczny zakup - brytyjska firma wydała 2,2 mld dol. w styczniu na RAPT Therapeutics i 950 mln dol. w lutym na 35Pharma. To również największe przejęcie dla spółki od ponad dekady.
GSK zaproponował cenę 124 dol. za każdą akcję Nuvalenta, co stanowi 40% premię wobec notowań giełdowych z dnia poprzedzającego ogłoszenie transakcji. Integracja kapitałowa ma zostać zakończona w III kw. 2026.
Transakcja wpisuje się w filozofię koncernu z Londynu, który ceni operacje w odniesieniu do aktywów zapewniających „wiele strzałów na bramkę” za jednym zamachem.
Źródło: GSK
Dzięki niej GSK wzbogacił swoje portfolio ukierunkowane na choroby nowotworowe, gdyż do już posiadanych gwiazd w postaci Jemperli (dostarlimab), Zejuli (niraparib) i Blenrepu (belantamab mafoditin) mogą dołączyć kolejne, tym razem z portfela przejmowanego od Nuvalenta.
Przejmowany podmiot jest „w przededniu” decyzji zatwierdzeniowych FDA dla dwóch cząsteczek przeznaczonych do terapii niedrobnokomórkowego raka płuc - dotyczy to zidesamtinibu i neladalkibu. Amerykański regulator wyznaczył daty wydania decyzji dopuszczeniowych odpowiednio na 18 września i 27 listopada br. Analitycy Truist Securities oszacowali sprzedaż szczytową dla obu kandydatów na leki na ok. 3,5 mld dol.
Przyznali przy tym, że GSK nie był najbardziej oczekiwanym strategicznym nabywcą omawianym w ich ostatnich rozmowach z inwestorami. Według nich przejęcie jest zgodne z ostatnimi priorytetami rozwojowymi brytyjskiego koncernu Big Pharmy i stwarza niskie ryzyko antymonopolowe przy ograniczonym nakładaniu się pipeline’ów obu firm.
- Parabilis Medicines pozyskał 633 mln dol. w ramach pierwszej oferty publicznej (IPO). Wskutek zgłoszonego przez inwestorów popytu rozmiar sprzedaży akcji podczas IPO uległ powiększeniu - z 25 mln akcji do 33,3 mln (bez zmiany przedziału cenowego 17-19 dol.). Dzięki tej decyzji Parabilis przeprowadziło największą tegoroczną ofertą publiczną, detronizując dotychczasowego lidera - Kailera Therapeutics, który pozyskał 625 mln dol.
Było to również największe IPO w ostatniej dekadzie. Kailera przejęła wcześniej palmę pierwszeństwa od Moderny, dewelopera szczepionek mRNA, które pomogły okiełznać pandemię COVID-19, a z oferty publicznej dostał 604 mln dol. od inwestorów.
Parabilis jest spółką na etapie klinicznym, którego głównym kandydatem na lek jest zolucatetide, cząsteczka badana pod kątem terapii kilku rodzajów guzów litych. Terapia dożylna, która ma na celu hamowanie interakcji między β-kateniną a rodziną czynników transkrypcyjnych T-komórkowych jest obecnie w fazie I/II terapii guzów desmoidalnych, a pieniądze z oferty publiczne pozwolą przesunąć ją do III fazy klinicznej, która rozpocznie się w pierwszej połowie 2027 r.
Portfolio firmy znalazło uznanie w oczach menedżerów Regeneron Pharmaceuticals, którzy zgodzili się zainwestować 75 mln dol. w równoległej ofercie prywatnej, wyceniającej akcje na równowartość 90% jednostkowej ceny z IPO. To spore wyróżnienie, ponieważ Regeneron jest 19. największą giełdową spółką biofarmaceutyczną świata z kapitalizacją rynkową 64 mld dol.
Debiut giełdowy Parabilis wypadł okazale, ponieważ kurs akcji na zamknięciu pierwszej sesji notowań zwyżkował o 61,2% w porównaniu z ceną z oferty publicznej.
Środa (10.6.2026)
- Sanofi zakończy badanie fazy III riliprubartu na przewlekłą zapalną polineuropatią demielinizacyjną po tym, gdy wyniki II fazy MOBILIZE wykazały, że prawdopodobnie cząsteczka nie zapewni wystarczającej skuteczności.
Decyzję podjęto na podstawie rekomendacji komitetu monitorującego dane, który wprawdzie nie stwierdził nowych problemów z bezpieczeństwem, lecz uznał, że lek prawdopodobnie nie osiągnie głównego punktu końcowego dotyczącego skuteczności. Sanofi zaznaczyło, że oceni przyszłość innych trwających badań z udziałem cząsteczki, w tym kolejnego badania fazy III VITALIZE.
Czwartek (11.6.2026)
- Takeda Pharmaceutical podała, że przyjmowany raz dziennie doustnie zasocitinib (TAK-279), wykazał statystyczną wyższość nad deucravacitinibem (Sotyktu) w zakresie głównego punktu końcowego, którym był odsetek całkowitego oczyszczenia skóry (PASI 100) po 16. tygodniach w łuszczycy. Ponad 35% pacjentów przyjmujących zasocitinib osiągnęło ten punkt, co oznacza ponad 2,5-krotnie lepszy wynik niż w grupie przyjmującej Sotyktu. To drugie badanie porównawcze, w którym zasocitinib pokonał dopuszczoną terapię - wcześniej dotyczyło to Otezli Amgenu.
Takeda weszła w posiadanie cząsteczki w wyniku przejęcia Nimbus Therapeutics, za którą zapłaciła z góry 4 mld dol. w 2022 r. Japoński koncern planuje wystąpić z wnioskiem o dopuszczenie zasocitinibu począwszy od roku obrotowego 2026. Gdyby firmie przyznano zgodę, to cząsteczka zaczęłaby konkurować m. in. z Sotyktu Bristol Myers Squibb. Sotyktu dostało zielone światło od FDA w 2022 r. i w zeszłym roku wygenerowało 291 mln dol. sprzedaży.
Piątek (12.6.2026)
- Jazz Pharmaceuticals ujawniła, że jej kandydat na lek w terapii raka płuca Zepzelca (lurbinectedin) nie osiągnął głównego celu poprawy całkowitego przeżycia w badaniu III fazy klinicznej.
Badanie LAGOON oceniało lurbinectedin samodzielnie lub łącznie z irynotekanem w porównaniu z topotekanem albo irynotekanem w grupie 724 pacjentów z nawrotowym, przerzutowym drobnokomórkowym rakiem płuc.
Spółka podkreśliła, że rezultat nie wpływa na zatwierdzenie Zepzelki w USA, które FDA przyznała jej w 2025 r. Ówczesna zgoda była oparta na badaniu IMforte, oceniającym cząsteczkę w leczeniu podtrzymującym pierwszej linii w połączeniu z atezolizumabem u chorych z rozsianą postacią drobnokomórkowego raka płuc.
- Novartis zaraportował, że delpacibart braxlosiran (del-brax) testowany w leczeniu dystrofii mięśniowej w odmianie twarzowo-łopatkowo-ramieniowej (FSHD), który został uzyskany w ramach przejęcia Avidity za 12 mld dol. w lutym 2026, wykazał obiecujące wyniki w badaniu I i II fazy klinicznej (FORTITUDE) - obniżył poziom biomarkerów KHDC1L (cDUX) oraz kinazy kreatynowej.
Szwajcarski koncern przyznał, że prowadzi rekrutację do badania III fazy. Triumf w niej mógłby oznaczać, że na rynku pojawi się pierwsza terapia modyfikująca przebieg FSHD.
"Dane kohorty biomarkerów FORTITUDE istotnie odzwierciedlają zaangażowanie celu i ochronę mięśniową w dalszej fazie, obserwowaną u del-brax we wcześniejszych kohortach z eskalacją dawki. Wyniki te potwierdzają schemat dawkowania stosowany w naszym badaniu fazy III i dostarczają dodatkowych dowodów na potencjał del-braksu do znaczącego wpływu na osoby z FSHD" - powiedział Nazem Atassi, globalny szef neurobiologii i rozwoju terapii genowej Novartisu.
- FDA wydała zgodę Sanofi dla Tzield (teplizumab-mzwv), ukierunkowanego na spowalnianie utraty własnej produkcji insuliny u dzieci w wieku od 8 do 17 lat, u których niedawno rozpoznano cukrzycę typu 1 w stadium 3. To pierwsza terapia modyfikująca przebieg choroby w tej grupie wiekowej pacjentów.
W listopadzie 2022 r. Tzield został dopuszczony do obrotu ze wskazaniem opóźnienia wystąpienia stadium 3 u dorosłych i dzieci w wieku co najmniej 8 lat, które miały stadium 2 choroby.
Lek jest przyjmowany w postaci codziennego wlewu podawanego przez 14 kolejnych dni, a wśród najczęściej zgłaszanych działań niepożądanych wystąpiły limfopenia, wysypka oraz leukopenia.
Obserwuj nas na  Google News
Google News
Chcesz być na bieżąco z wieściami z naszego portalu? Obserwuj nas na Google News!

Reklama
Komentarze opinie
Podziel się swoją opinią
Twoje zdanie jest ważne jednak nie może ranić innych osób lub grup.





Reklama
Reklama




Reklama
Reklama
Reklama
Najnowsze wiadomości
- 14/06 PoZdroweek 24/2026: GSK przejmie Nuvalent Bio za prawie 11 mld dol.
- 14/06 Mazowiecki Szpital Specjalistyczny z gigantycznym wsparciem. 32,5 mln zł z KPO na cyfryzację i kardiologię
- 14/06 „Medstudent” Medycznym Słowem Roku 2026. Takim językiem posługują się dziś studenci kierunków medycznych
- 14/06 Czemu mężczyzna nie biega? Cztery najważniejsze przyczyny
- 14/06 Uniwersytet Medyczny w Lublinie liderem w regionie. Awansował do czołówki Rankingu Polskich Uczelni 2026
- 14/06 Nowa szczepionka przeciw fentanylowi daje obiecujące wyniki. Może blokować nielegalne pochodne narkotyku
- 14/06 Polacy i Estończycy odkryli błąd mogący zniekształcać wyniki badań mikrobiomu jelitowego
- 14/06 Ponad 60 mln zł na kształcenie lekarzy. Uniwersytet Opolski inwestuje w VR i symulatory medyczne
- 14/06 USK Nr 4 w Lublinie szuka pacjentek z endometriozą. Badanie ma pomóc w stworzeniu nowych metod leczenia
- 13/06 Bezsenność i lęk u seniorów pod kontrolą dzięki aplikacji. Potwierdzono skuteczność terapii online
- 13/06 Pierwsza miesiączka może zaskoczyć już 8-latkę. Przed wakacjami Warto porozmawiać z córką o dojrzewaniu?
- 13/06 Grupa Odyseusza z nagrodą Psychiatryczna Innowacja Roku 2026
- 13/06 Szpital w Sosnowcu zakończył cyfrową transformację za ponad 14 mln zł
- 13/06 Uniwersytet Medyczny w Łodzi z nową bazą dydaktyczną za 190 mln zł
- 13/06 Kardiolodzy: mało inwazyjne zabiegi implantacji zastawki aortalnej nie powinny być limitowane
- 13/06 Ministerstwo Zdrowia ostro o prezydencie. Będzie drugie podejście z ustawą
- 13/06 130 mln zł na krwiodawstwo w Polsce. Ministerstwo Zdrowia ogłosiło nowy program do 2032 roku
- 12/06 Global Cardio-Oncology Summit 2028 w Warszawie. NIO doceniony za rozwój kardioonkologii
- 12/06 „66 proc. dzieci boi się rezonansu magnetycznego”. Polski szpital pokazuje, jak zmniejszyć stres najmłodszych pacjentów o 43 proc.
- 12/06 Turystyka medyczna. Tam Polacy jeżdżą leczyć zaćmę
Komentarze